
Abstract
In this work, we report the use of ascorbic acid-capped silver nanoparticles (AA–Ag NPs) as a probe for selective colorimetric detection of glutathione (GSH) in aqueous solution. This detection system was based on the GSH-induced aggregation of AA–Ag NPs, resulting in drastic changes in the absorption spectra and color of the AA–Ag NPs system. The GSH-induced AA–Ag NPs aggregation was confirmed by UV–visible spectrometry, dynamic light scattering (DLS) and transmission electron microscopic (TEM) techniques. Under optimal conditions, this method exhibited good linearity over the concentration ranges from 5.0 to 50 µM, with the limit of detection 2.4 × 10−7 M. This method was successfully applied to detect GSH in the presence of other biomolecules, which confirms that this probe can be used for the detection of GSH in real samples.
Similar content being viewed by others

Introduction
Glutathione (GSH, γ-glutamylcysteinylglycine) is a primary non-protein sulfhydryl in most of the cells. It contains an unusual peptide linkage between γ-carboxyl group of Glu side chain and amino group of cysteine (Valko et al. 2007). It plays a key role in maintaining redox homeostasis in cells and acts as an antioxidant for keeping the thiol group of cysteine in proteins (Dröge 2002). The reduced glutathione/oxidized glutathione ratio is used to evaluate oxidative stress in cells. It serves as a cofactor for glutathione peroxidase in the decomposition of hydrogen peroxide or organic peroxides and for glyoxalase 1 in the detoxification of methylglyoxal and other α-oxo-aldehydes (Metzler 2004). Furthermore, it plays a significant role in studying various diseases such as HIV, diabetes, liver damage, heart diseases, mild cognitive impairment, edema, pneumonia and skin lesions (Droge and Holm 1997; Staal 1998; Micke et al. 2001; Julius et al. 1994; Bray and Taylor 1994). Therefore, it is highly desirable to develop a new sensitive, fast and practical method for the detection of GSH in biological samples. Although conventional analytical tools including electrospray ionization mass spectrometry (ESI–MS) (Kailasa et al. 2012), matrix-assisted laser desorption/ionization mass spectrometry (MALDI–MS) (Wan et al. 2013), high-performance liquid chromatography (HPLC) (Casey et al. 2002), fluorescence spectrometry (Shao et al. 2010) and electrochemistry (Safavi et al. 2009) could meet the demand for the sensitive and selective detection of GSH in biological samples, all these methods are expensive, time-consuming and require tedious sample preparations. In this frame, it is necessary to develop a facile and sensitive method for the analysis of GSH in various samples.
The integration of nanotechnology with analytical chemistry is becoming increasingly important for the development of miniaturized analytical tools for ultrasensitive detection of various target analytes including biomolecules and inorganic and organic molecules (Kailasa and Wu 2013, Kailasa et al. 2013; Vilela et al. 2012). Metallic nanoparticles (Au and Ag NPs) have attracted extensive research interest as a promising colorimetric probe for sensing different target analytes in the past two decades, due to their easy preparation and unique optical properties [surface plasmon resonance absorption (SPR) and resonance light scattering] (Jiang et al. 2011). As a result, many functionalized Ag NPs have been used as colorimetric sensors for the detection of a wide variety of molecules including organic (Tashkhourian et al. 2011; Zhang et al. 2010), inorganic (Mehta et al. 2013; Modi et al. 2014; Yao et al. 2010) and biomolecules (Ravindran et al. 2011; Yuan et al. 2013). These methods are based on the analytes-induced aggregation of Ag NPs, resulting in changes in both color and absorption spectrum. These analytes-induced aggregations are due to the interactions between surfaces of Ag NPs and analytes, confirming that the surface modification of NPs is very important to improve the stability and sensing ability of Ag NPs. So far, the use of ascorbic acid-functionalized Ag NPs as a colorimetric sensor for the detection of GSH has been unexplored. Herein, we report a novel colorimetric method for the detection of GSH using ascorbic acid-functionalized Ag NPs as a colorimetric probe.
Materials and methods
Chemicals and materials
Silver nitrate, sodium borohydride, ascorbic acid, sodium acetate and glutathione were purchased from Sigma-Aldrich, USA. Threonine, histidine, glutamic acid, serine, lysine HCl, cysteine, tyrosine and tryptophan were purchased from SRL Chemicals. Milli-Q-purified water was used for the sample preparations.
Synthesis of AA–Ag NPs
Ag NPs were prepared according to the method in the literature (Pinto et al. 2010) with minor modifications. Briefly, 5 mL of 0.25 mM silver nitrate was taken into a 50-mL reaction flask that contained 5 mL of 0.1 mM ascorbic acid and then stirred for 30 s. To this, 0.5 mL of 1.5 mM NaBH4 was added dropwise and stirred for 10 min, resulting in a color change from colorless to light yellow, which confirms the formation of AA–Ag NPs. Supporting information of Scheme 1 shows the schematic representation for the preparation of AA–Ag NPs.
Detection of GSH
For detection of glutathione using AA–Ag NPs, different concentrations of GSH (5.0–50 µM, 0.1 mL) were added separately into sample vials that contained 1 mL of AA–Ag NPs at sodium acetate buffer pH 7.0. As a result, the color of AA–Ag NPs was changed from bright yellow to orange-brown color, indicating that the aggregation of AA–Ag NPs was induced by GSH. Subsequently, their absorption spectra were measured by UV–visible spectrometer.
Instrumentation
Absorption spectra were recorded on a Maya Pro 2000 spectrophotometer, Ocean Optics, USA. Dynamic light scattering (DLS) data were measured using Zetasizer Nano ZS90 (Malvern, UK). Fourier transform-infrared (FT-IR) spectra were recorded using FT-IR 8400S (Shimadzu, Japan) with KBr powder. Transmission electron microscopy (TEM) images were taken using CM200 (Philips, Netherlands).
Results and discussion
Characterization of AA–Ag NPs
The synthesized AA–Ag NPs were characterized by FT-IR, DLS, TEM and UV–visible spectroscopy. To improve the sensing ability of Ag NPs, we optimized the concentrations of NaBH4 as a reducing agent and AA as a capping agent. As shown in Fig. 1a and supporting information of Fig. S1a, the best absorption spectrum of AA–Ag NPs was obtained using 1.5 mM of NaBH4 as a reducing agent and 0.1 mM of AA as a capping agent. It can be observed that there is no obvious change in the SPR band and a characteristic SPR band at 397 nm exists, which suggest that the AA–Ag NPs have the optical property of the original Ag NPs. We also studied the effect of reaction temperature on the formation of Ag NPs. It was noticed that the maximum absorbance (at 397 nm) and intense yellow color were observed by performing the reaction at room temperature (supporting information of Fig. S1b). Supporting information of Fig. S2 shows the FT-IR spectra of pure AA and AA–Ag NPs. The peaks at 3,526 and 3,412–3,216 cm−1 were not observed in the FT-IR spectrum of AA–Ag NPs, which confirms the oxidation of hydroxy groups in AA. The pure AA spectrum showed peaks at 3,212–3,626 cm−1, which corresponded to stretching of the –OH groups. The peaks at 3,030 and 2,879 cm−1 corresponded to the asymmetric and symmetric vibrations of –CH2 group. The carbonyl group (C=O) stretching was observed at ~1,760 cm−1 and the vibration frequency of C=C was observed at ~1,670 cm−1. It can be observed that the –OH group stretching disappeared at 3,212–3,626 cm−1, which indicates that AA molecules successfully interacted with Ag NPs. Figure 2a and c shows the DLS and TEM image of AA–Ag NPs, which indicate that AA–Ag NPs are well dispersed in water with uniform size. The average diameter of AA–Ag NPs is 2.5 nm.

a UV–visible spectra of Ag NPs using ascorbic acid (0.01–0.25 mM) as a capping agent. b UV–visible spectra of AA–Ag NPs at different sodium acetate buffer pH values from 2 to 10

DLS of a AA–Ag NPs and b GSH-induced aggregation of AA–Ag NPs. TEM images of c AA–Ag NPs and d GSH-induced aggregation of AA–Ag NPs
AA–Ag NPs as a probe for colorimetric sensing of GSH
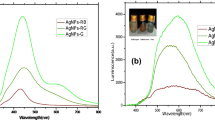
To investigate AA–Ag NPs as a colorimetric sensor, various biomolecules (threonine, histidine, glutamic acid, serine, lysine HCl, cysteine, tyrosine, tryptophan and GSH, 0.1 mL, 1 mM) solutions were added into 1 mL of AA–Ag NPs solutions with sodium acetate buffer pH 7.0. As shown in Fig. 3, the characteristic SPR peak of AA–Ag NPs at 397 nm is redshifted to 468 nm only by the addition of GSH, resulting in a color change from yellow to orange-brown, which confirms that the aggregation of AA–Ag NPs was induced by GSH. To investigate the effect of pH condition on the absorption spectra of AA–Ag NPs, we measured UV–visible spectra of AA–Ag NPs with sodium acetate buffer (pH 2.0 to 10.0) without GSH. As shown in Fig. 1b, the UV–visible spectra of AA–Ag NPs are drastically changed at lower pH of 2.0–4.0, which is due to the surface neutralization of AA–Ag NPs. At the same time, UV–visible absorption spectrum of AA–Ag NPs remained the same at pH 7.0, which suggests that the color and absorption spectrum of AA–Ag NPs did not influence sodium acetate buffer pH at pH 7.0. To investigate the effect of pH on GSH-induced aggregation of AA–Ag NPs, we studied the UV–visible spectra of AA–Ag NPs upon the addition of GSH with sodium acetate buffer pH in the range of 2.0–10.0 (Fig. 4). With sodium acetate buffer pH 7.0, the characteristic SPR band of AA–Ag NPs at 397 nm is redshifted toward longer wavelength (468 nm), resulting in a color change from yellow to orange-brown, which confirms the aggregation of AA–Ag NPs induced by GSH. AA–Ag NPs (AA—pKa 4.10) surfaces and GSH (pI = 5.90) have exhibited negative and positive charges, which is due to their pI values. As a result, there is strong electrostatic interaction between AA–Ag NPs and GSH. Furthermore, GSH strongly induced the aggregation of AA–Ag NPs because of the stronger affinity of GSH toward AA–Ag NPs, due to the multidentate anchoring groups (e.g., –SH, –NH2 and –COO−) of GSH. Therefore, GSH effectively induced the aggregation of AA–Ag NPs via multiple interactions (electrostatic and covalent), resulting in changes in both UV–visible spectra and color of the AA–Ag NPs. To confirm the GSH-induced AA–Ag NPs aggregation, we measured the DLS and TEM of AA–Ag NPs in the presence of GSH (Fig. 2b, d). It can be observed that AA–Ag NPs sizes increased to 619.5 nm, which confirms that the aggregation of AA–Ag NPs was induced by GSH. Supporting information of Fig. S3 shows the absorbance ratio at A468nm/A397nm that can describe the degree of aggregation ability of GSH with AA–Ag NPs. Based on the above results, AA–Ag NPs acted as a selective colorimetric probe for sensing of GSH in aqueous solution.

UV–visible spectra of a AA–Ag NPs in the presence of GSH over different amino acids (threonine, lysine, serine, cysteine, histidine, glutamic acid, tyrosine, tryptophan) and b photographic image of AA–Ag NPs in the presence of various amino acids

a UV–visible spectra of AA–Ag NPs by the addition of GSH at sodium acetate buffer pH values from 2.0 to 10.0. b Photographic image of the corresponding solutions
To construct the calibration graph for GSH analysis, we measured the UV–visible spectral changes of AA–Ag NPs by the addition of different concentrations of GSH from 5.0 to 50 µM (Fig. 5a). It can be observed that the new SPR peak intensity was gradually increased with increase in the concentration of GSH, which confirms that the degree of GSH-induced AA–Ag NPs aggregation was increased with increase in the concentration of GSH up to 50 µM, and resulted in a redshift in wavelength (from 397 to 468 nm) and a color change from yellow to orange-brown (Fig. 5b). Under optimal conditions, a linear relationship (R2 = 0.997) exists between the absorption ratio (A468nm/A397nm) and the concentration of GSH over the range of 5.0 and 50 µM with the limit of detection 2.4 × 10−7 M (supporting information of Fig. S4). To evaluate the sensing capability of AA–Ag NPs in the presence of other biomolecules, we studied the absorption spectra of AA–Ag NPs upon the addition of GSH in the presence of difference molecules (threonine, histidine, glutamic acid, serine, lysine HCl, cysteine, tyrosine, and tryptophan, 0.1 mL, 1 mM) and the obtained spectra are shown in supporting information of Fig. S5. It was noticed that the AA–Ag NPs system successfully detected the GSH molecules in the presence of other similar molecules, which represents that the developed system could not produce any wrong signal in the presence of other similar analytes. These results suggest that AA–Ag NPs-based sensor exhibited high selectivity to detect GSH in the presence of other threonine, histidine, glutamic acid, serine, lysine HCl, cysteine, tyrosine and tryptophan molecules.

a UV–visible spectra of AA–Ag NPs by the addition of GSH in the range of 5.0–50 µM. b Photographic image of the corresponding solutions
Conclusions
In summary, we have developed a facile and selective colorimetric method for detection of GSH using AA–Ag NPs. The GSH-induced AA–Ag NPs aggregation results in a noticeable redshift (from 397 to 468 nm) in the UV–visible absorption spectrum and a visible color change from yellow to orange-brown, which allow the detection of GSH with a detection limit of 2.4 × 10−7 M. The AA–Ag NPs acted as a colorimetric sensor for the detection of GSH in the presence of other similar molecules. This method provides a simple platform for the detection of GSH in biological samples with high selectivity and sensitivity.
References
Bray TM, Taylor CG (1994) Enhancement of tissue glutathione for antioxidant and immune functions in malnutrition. Biochem Pharmacol 47:2113–2123
Casey W, Anderson S, Fox T, Dold K, Colton H, Morgan K (2002) Transcriptional and physiological responses of HepG2 cells exposed to diethyl maleate: time course analysis. Physiol Genomics 8:115–122
Dröge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Droge W, Holm E (1997) Role of cysteine and glutathione in HIV infection and other diseases associated with muscle wasting and immunological dysfunction. FASEB J 11:1077–1089
Jiang Z, Zhou L, Liang A (2011) Resonance scattering detection of trace melamine using aptamer-modified nanosilver probe as catalyst without separation of its aggregations. Chem Commun 47:3162–3164
Julius M, Lang CA, Gleiberman L, Harburg E, Difranceisco W, Schork A (1994) Glutathione and morbidity in a community-based sample of elderly. J Clin Epidemiol 47:1021–1026
Kailasa SK, Wu H-F (2013) Surface-assisted laser desorption-ionization mass spectrometry of oligosaccharides using magnesium oxide nanoparticles as a matrix. Microchim Acta 180:405–413
Kailasa SK, Hasan N, Wu H-F (2012) Identification of multiply charged proteins and amino acid clusters by liquid nitrogen assisted spray ionization mass spectrometry. Talanta 97:539–549
Kailasa SK, Cheng K-H, Wu H-F (2013) Semiconductor nanomaterials-based fluorescence spectrometric and MALDI mass spectrometric approaches to proteome analyses. Materials 6:5763–5795
Mehta VN, Mungara AK, Kailasa SK (2013) Dopamine dithiocarbamate functionalized silver nanoparticles as colorimetric sensors for the detection of cobalt ion. Anal Methods 5:1818–1822
Metzler DE (2004) Biochemistry, the chemical reactions of living cells, 2nd edn. Elsevier, Academic Press
Micke P, Beeh KM, Schlaak JF, Buhl R (2001) Oral supplementation with whey proteins increases plasma glutathione levels of HIV-infected patients. Eur J Clin Investig 31:171–178
Modi RP, Mehta VN, Kailasa SK (2014) Bifunctionalization of silver nanoparticles with 6-mercaptonicotinic acid and melamine for simultaneous colorimetric sensing of Cr3+ and Ba2+ ions. Sensor Actuat B Chem 195:562–571
Pinto VV, Ferreira MJ, Silva R, Santosc HA, Silva F, Pereirab CM (2010) Long time effect on the stability of silver nanoparticles in aqueous medium: effect of the synthesis and storage conditions. Colloids Surf A Physicochem Eng Aspects 364:19–25
Ravindran A, Mani V, Chandrasekaran N, Mukherjee A (2011) Selective colorimetric sensing of cysteine in aqueous solutions using silver nanoparticles in the presence of Cr3+. Talanta 85:533–540
Safavi A, Maleki N, Farjami E, Mahyari FA (2009) Simultaneous electrochemical determination of glutathione and glutathione disulfide at a nanoscale copper hydroxide composite carbon ionic liquid electrode. Anal Chem 81:7538–7543
Shao N, Jin J, Wang H, Zheng J, Yang R, Chan W, Abliz Z (2010) Design of bis-spiropyran ligands as dipolar molecule receptors and application to in vivo glutathione fluorescent probes. J Am Chem Soc 132:725–736
Staal FJT (1998) Glutathione and HIV infection: reduced reduced, or increased oxidized? Eur J Clin Investig 28:194–196
Tashkhourian J, Hormozi MR, Khodaveisi J, Dashti R (2011) A novel photometric glucose biosensor based on decolorizing of silver nanoparticles. Sens Actuators B 158:185–189
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J (2007) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Bio 39:44–84
Vilela D, González MC, Escarpa A (2012) Sensing colorimetric approaches based on gold and silver nanoparticles aggregation: chemical creativity behind the assay. a review. Anal Chim Acta 751:24–43
Wan D, Gao M, Wang Y, Zhang P, Zhang X (2013) A rapid and simple separation and direct detection of glutathione by gold nanoparticles and graphene-based MALDI-TOF-MS. J Sep Sci 36:629–635
Yao Y, Tian D, Li H (2010) Cooperative binding of bifunctionalized and click-synthesized silver nanoparticles for colorimetric Co2+ sensing. ACS Appl Mater Interfaces 2:684–690
Yuan X, Tay Y, Dou X, Luo Z, Leong DT, Xie J (2013) Glutathione-protected silver nanoclusters as cysteine-selective fluorometric and colorimetric probe. Anal Chem 85:1913–1919
Zhang YF, Li BX, Chen XL (2010) Simple and sensitive detection of dopamine in the presence of high concentration of ascorbic acid using gold nanoparticles as colorimetric probes. Microchim Acta 168:107–113
Acknowledgments
This work was financially supported by the Department of Science and Technology, Government of India, India for Inspire Ph.D programme. The authors would like to thank Prof. Murthy and Mr. Chetan Patel for their help with DLS measurements. We also thank the Department of Science and Technology for providing Maya Pro 2000 spectrophotometer under the Fast-Track Young Scientist Scheme (2011–2014).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
D’souza, S.L., Pati, R. & Kailasa, S.K. Ascorbic acid-functionalized Ag NPs as a probe for colorimetric sensing of glutathione. Appl Nanosci 5, 747–753 (2015). https://doi.org/10.1007/s13204-014-0371-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13204-014-0371-9