
Abstract
Triple negative breast cancer (TNBC) is an aggressive breast cancer subtype that lacks estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) amplification. Due to the absence of these receptors, TNBC does not respond to traditional endocrine or HER2-targeted therapies that improve patient prognosis in other breast cancer subtypes. TNBC has a poor prognosis, and currently, there are no effective targeted therapies. Some TNBC tumors express androgen receptor (AR) and may benefit from AR-targeted therapies. Here, we review the literature on AR in TNBC and propose that TNBC be further sub-classified as either AR+ TNBC or quadruple negative breast cancer since targeting AR may represent a viable therapeutic option for a subset of TNBC.
Similar content being viewed by others


Introduction
Triple negative breast cancer (TNBC), a term first published in 2005 [1], is defined by negative clinical testing for estrogen receptor (ERα), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) amplification. Because it lacks these receptors, TNBC is unresponsive to traditional estrogen pathway-directed endocrine therapies or HER2-targeting therapies. Currently, there are no targeted therapies for TNBC, and chemotherapy remains the best therapeutic option. However, upon recurrence of chemoresistant disease, effective therapeutic options are limited. Indeed, TNBC constitutes 15–20 % of newly diagnosed invasive breast carcinomas and has the lowest 5-year survival rate compared to other breast cancer (BC) subtypes. A study of over 1600 women found that women with TNBC have a peak risk of recurrence between 1 and 3 years, an increased likelihood of distal recurrence, and a majority of deaths occurring in the first 5 years compared to other BC subtypes [2]. Overall BC outcomes have greatly improved as a result of early diagnosis, however TNBC often presents between mammograms [2].
Although TNBC lacks hormone receptors traditionally associated with BC, both molecular and immunohistochemical analyses demonstrate that a subset of TNBC expresses the androgen receptor (AR). Emerging data suggest that AR significantly influences breast cancer gene expression profiles and affects tumorigenic properties of TNBC. Development of new generation anti-androgens for the treatment of prostate cancer has led to renewed interest in hormonal therapy targeting AR in the subset of AR+ TNBC and constitutes a novel therapeutic option that could improve prognosis with few side effects. Herein, we discuss the role of AR in the biology of TNBC in preclinical models and review the clinical data on the efficacy of targeting AR in TNBC. Based on these data, we propose that testing for AR should become the standard clinical practice and that TNBC be further defined as either AR+ TNBC or “quadruple negative” disease to emphasize the utility of AR as a viable therapeutic target in AR+ breast cancer.
AR Expression in TNBC
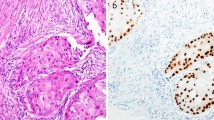
Multiple studies report nuclear AR expression in TNBC patient specimens [3–11]. Importantly, nuclear AR staining is indicative of active receptors, since AR translocates to the nucleus upon binding ligand. The percentage of TNBC with nuclear AR expression (as detected by IHC) ranges from 12–55 % depending on the study. The large range of AR expression may reflect differences in AR antibodies used, antigen retrieval methods, and the criteria for positive AR expression (either 1 or 10 % positivity). In two prospectively conducted clinical trials, rates of AR+ disease ranged from 12 % (AR ≥10 %, DAKO) [10] to 55 % (AR ≥10 %, DAKO and Ventana) [11]. Studies comparing AR expression in primary versus metastatic disease found that AR is frequently retained in metastatic samples from patients with AR+ primary tumors [12, 13]. Reports of AR expression in TNBC patient samples are summarized in Table 1. To date, there is no standardized, optimized assay to assess AR expression in TNBC. As we move toward classification of AR+ and quadruple negative disease, standardization of AR IHC is of high priority. Interestingly, the percent of AR+ cells alone may not be sufficient to identify patients who will benefit from AR-targeted therapy. For example, the genomic biomarker reported by Traina et al. predicted patients who would benefit from enzalutamide (Enza) despite having low AR expression by IHC [14]. AR expression alone was not significantly associated with patient outcome. Thus, a combination of AR expression by IHC ≥1 % and genomic biomarker expression may best identify the cohort of AR+ TNBC patients who will benefit from AR-targeted therapy.
Across all subtypes of BC, AR expression is associated with a better overall survival and disease-free survival irrespective of co-expression of ERα in breast cancer [15]. Within TNBC, the prognostic significance of AR is controversial (reviewed in [16]) as AR expression has been associated with both a good and bad prognosis in multiple studies. As AR is expressed in normal mammary epithelial cells (see primary data in [17]), it is likely associated with a more well-differentiated, less aggressive tumor. Indeed, AR+ TNBC has a lower Ki-67 index than AR− TNBC [3] and may therefore be less responsive to chemotherapy. This is supported by the lower pathologic complete response (pCR) rate following chemotherapy of TNBC tumors with gene expression profiles enriched in AR signaling pathways [18] compared to other TNBC subtypes. Just as ERα confers a good prognosis, but is an effective therapeutic target in ER+ breast cancer [19–22], AR may similarly confer an overall better prognosis since it is expressed in slower growing tumors, yet serve as an effective therapeutic target in a subset of TNBC that are dependent on or driven by this receptor.
In TNBC tumor cohorts, studies identified correlations between AR protein positivity and other proteins of biological importance. AR protein expression is positively correlated with aldehyde dehydrogenase 1 (ALDH1) [23], an enzyme associated with stem-like cells in breast cancer, as well as 17β-hydroxysteroid dehydrogenase 5 (17βHSD5) and 5 alpha-reductase type 1 (5αR1), enzymes involved in androgen synthesis [24]. Conversely, AR protein expression is negatively correlated with the L1 cell adhesion molecule (L1CAM) [25] and tight junction protein Claudin 4 (CLDN4) [26]. These studies raise the interesting possibility that AR may promote a stem-like or mesenchymal phenotype in TNBC, an observation consistent with the lower Ki67 staining associated with AR+ TNBC.
TNBC Lacks AR Mutations but Expresses AR Splice Variants
The Cancer Genome Atlas (TCGA) sequencing data revealed 2 patients with single missense mutations among 93 TNBCs analyzed (2.2 % mutation rate). One mutation, D865E, localized to the ligand-binding domain of AR and the other, L638M, localized to a domain for which a specific function have not been ascribed. Neither mutation contributes to a reported or predicted functional alteration of AR. By FISH analysis of 99 BC and normal breast tissues, AR gene amplification was not found [27]. As tissue from clinical trials targeting AR in TNBC becomes available, amplification or mutations in AR may be identified as a result of AR-targeted therapy.
AR splice variant mRNA is expressed in BC tissues and cell lines [28, 29]. An AR variant without exon 3 (Δ3AR) that lacks the second zinc finger of the DNA binding domain was identified in BC, but not normal, tissues [28]. Hu et al. also reported low levels of AR variant expression in normal breast tissue with some expression of AR variant AR45, which is truncated at the N-terminus [29]. AR45 lacks exon 1 and is associated with decreased AR activity in prostate cancer [30]. TNBC cell lines MDA-MB-453 and MDA-MB-231 express AR45 and AR variant 3, respectively [29]. AR variant 3, which lacks a ligand binding domain, is ligand independent and constitutively active in prostate cancer [31]. Additional studies are needed to verify AR variant expression in TNBC tumor specimens at the protein level, particularly after treatment with AR-targeted therapies.
AR and Hormone-Regulated Genes Define a Subtype of TNBC
Molecular profiling experiments have redefined BC subtypes to take into account the strong effects of AR, which is expressed across BC subtypes [32–34]. In a study examining mRNA expression patterns in over 500 breast tumors representing all BC subtypes, Guedj et al. identified an AR-regulated gene cluster as one of three key gene clusters that define global BC expression patterns [32]. The AR-associated gene cluster included a molecular apocrine AR+/PR−/ER− subgroup (mApo, comprised of tumors with and without HER2 amplification) as well as a basal-like AR−/ER−/PR− subgroup (BasL) [32]. Whereas the mApo subgroup was enriched in immune signaling pathways, the BasL subgroup was enriched in DNA replication and repair signaling. The expression profile of the mApo, AR+/ER−/PR− subgroup is similar to the previously described “molecular apocrine” group of tumors enriched with expression of ER-regulated genes, despite being ER− [33, 34]. It is proposed that in these tumors, AR promotes transcription of many of these classically ER-regulated genes.
More recently, expression analysis of over 500 TNBC patient samples identified significant heterogeneity that includes a subtype with a “luminal AR” gene signature as well as “basal-like 1,” “basal-like 2,” “immunomodulatory,” “mesenchymal-like,” and “mesenchymal stem-like” subtypes [35]. The luminal AR (LAR) subtype was so termed because it has a molecular expression profile similar to ER+ breast cancers. Gene ontologies defining the LAR subtype were enriched in hormonally regulated pathways including steroid synthesis and androgen/estrogen metabolism. Importantly, the LAR subtype was associated with a poor patient prognosis suggesting that this subtype may be less responsive to chemotherapy. Masuda and colleagues also compared pCR rates following neoadjuvant chemotherapy of the TNBC subtypes and found that the pCR rates differed by subtype [18]. The basal-like 1 subtype had the highest pCR rate whereas the LAR and basal-like 2 subtypes had the lowest pCR rates. A low Ki-67 index associated with reduced proliferation may account for the low pCR rate among LAR tumors.
Subsequent TNBC profiling studies corroborated the existence of a LAR TNBC subtype. Recently, Burstein and colleagues examined 198 TNBC tumors and identified four distinct subtypes including LAR with the subtype specific marker mucin 1 (MUC1) [36]. Similarly, in a study of 107 TNBC, Jezequel et al. identified three subtypes including LAR [37]. Identification of an AR signaling-based subgroup of TNBC tumors by multiple independent studies further supports the need to distinguish quadruple negative breast cancer and AR+ disease in the clinic to optimize therapy and improve patient outcomes.
Molecular Biology of AR in the Luminal AR TNBC Subtype
LAR tumors exhibit a tenfold higher AR protein expression than non-LAR subtypes [35]. Xenograft experiments with LAR TNBC cell lines demonstrated greater sensitivity to the AR antagonist bicalutamide compared to non-LAR xenografts. However, bicalutamide is known to have partial agonist effects [38], and prostate cancer patients who acquire resistance to bicalutamide are often responsive to the next generation antagonist Enza, which has higher affinity for AR and attenuates DNA binding [39], while bicalutamide allows nuclear translocation and DNA binding. Indeed, non-LAR, AR+ SUM159PT and HCC1806 xenografts that were insensitive to bicalutamide [35] were sensitive to Enza [3], suggesting that response to pure AR antagonists may not be limited to the LAR TNBC subtype.
LAR cell lines have a high frequency of phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutations [40]. The combination of PI3K/mTOR and AR inhibitors showed additive growth inhibitory effects in vitro. Additive effects of bicalutamide with PI3K inhibitor GDC-0941 or PI3K/mTOR inhibitor GDC-0980 were also observed in MDA-MB-453 and CAL-148 LAR xenografts [40]. In a comparison of global DNA-binding events, Robinson et al. found that AR binding in the LAR TNBC cell line MDA-MB-453 was more similar to ER binding in the ER+ breast cancer cell line (MCF7) than AR binding in a prostate cancer cell line (LNCaP) [41]. Thus, in the absence of ER, AR may function similarly to ER, accounting for the luminal transcriptome of this subtype. AR binding further required the forkhead box protein A1 (FOXA1) pioneer factor.
Most research on the LAR subtype has utilized the MDA-MB-453 cell line. This cell line is classified as LAR by gene expression profiling [35], is strongly positive for AR by western blot and IHC, and is very responsive to new anti-androgens [42, 43]. However there is discrepancy with regard to HER2 amplification and overexpression status, rendering the status of this cell line as true TNBC debatable. Reported HER2 gene amplification rates as measured by FISH range from 1.08–2.39 [44–47]. Our own internal testing of MDA-MB-453 from two separate sources revealed FISH scores varying between 1.43 and 2.83. Clinically, positive HER2 amplification is defined as a FISH ratio (HER2-to-CEP17) higher than 2.2 or HER2 gene copy greater than 6 [48]. Studies have also shown that some MDA-MB-453 cell lines are innately non-responsive to trastuzumab [49, 50]. In summary, amplification of HER2 is present in some MDA-MB-453 cell lines and reliance on the HER2 receptor is likely dependent on the HER2 amplification levels of MDA-MB-453 cell lines that have drifted in various laboratories. Thus, in order to classify MDA-MB-453 as a TNBC line, research groups should test their MDA-MB-453 cell lines for HER2 amplification status and perhaps this line may represent a TNBC that is HER2-enriched [51]. MDA-MB-453 also has an AR mutation in the ligand binging domain that decreases receptor activity [52] and may render it a unique model of AR + TNBC, although it has high AR protein levels and is very responsive to anti-androgens in vitro and in vivo [42]. Further studies using additional LAR cell lines and patient-derived xenografts may facilitate a better characterization of the biology of this TNBC subtype.
Molecular Biology of AR in Non-Luminal AR TNBC Subtypes
Although AR is most highly expressed in LAR tumors, AR is expressed in non-LAR subtypes [3, 53]. Cell lines representing non-LAR and AR+ TNBC subtypes, including mesenchymal stem-like, mesenchymal-like and basal-like, exhibit decreased anchorage-independent growth when treated with the AR antagonist Enza [3]. AR knockdown and treatment with Enza decreased proliferation and increased apoptosis as measured by cleaved caspase-3. Migration and invasion were also inhibited by AR knockdown and treatment with Enza was associated with changes in cellular morphology from stellate to round. Additionally, basal-like HCC1806 and mesenchymal stem-like SUM159PT xenografts, with relatively low AR expression, treated with Enza exhibited decreased viability and increased necrosis. Response to Enza in non-LAR xenografts with relatively low AR expression provides promising preclinical data that TNBC patients with relatively low AR expression may also benefit from anti-androgen therapy. Indeed, results of the TBCRC011 trial demonstrate that some non-LAR TNBC benefit from treatment with bicalutamide [54]. This emerging clinical data in support of anti-androgen therapy for the treatment of AR+ TNBC, regardless of molecular subtype, further underscores the possible utility of classifying TNBC as AR+ or quadruple negative breast cancer.
Amphiregulin (AREG), an epidermal growth factor receptor (EGFR) ligand, was identified as an AR-regulated gene and exogenous AREG partially rescues decreased proliferation, migration, and invasion resulting from AR inhibition in vitro [3], suggesting that activation of EGFR is one mechanism by which AR affects TNBC biology. An association between AR and EGFR activity in TNBC was also identified by Cuenca-Lopez et al. [55]. In TNBC tumor lysates, AR protein expression significantly correlated with phosphorylation of EGFR and platelet-derived growth factor receptor beta (PDGFRβ). Combined treatment of the AR antagonist bicalutamide with inhibitors of EGFR, PDGFRβ, and Erk1/2 exhibited an additive anti-proliferative effect and decreased AR protein expression in non-LAR TNBC cell lines [53].
Graham and collaborators found that cross talk between AR and zinc finger E-box binding homeobox 1 (ZEB1), a transcription factor associated with epithelial-to-mesenchymal transition, influences migration of MDA-MB-231 and MDA-MB-435 cells [56]. ZEB1 can bind the AR promoter and ZEB1 knockdown decreases AR transcript, protein, and downstream AR targets. Likewise, treatment with the AR antagonist bicalutamide decreased ZEB1 protein expression. In the presence of ZEB1 knockdown, bicalutamide decreased migration. By tissue microarray, ZEB1 and AR expression were positively correlated in ER−/PR− BC tissues [56].
Clinical Efficacy of AR Inhibitors in AR+ TNBC
Recent clinical trials evaluating the efficacy of AR antagonists in AR+ TNBC are promising. A phase II trial of bicalutamide in AR+/ER−/PR− metastatic breast cancer found a 6-month clinical benefit rate (CBR) of 19 % [10]. The median progression free survival (PFS) was 12 weeks and bicalutamide was well-tolerated. While bicalutamide permits AR nuclear localization and disrupts the major coactivator binding surface on AR [57], Enza induces a conformational change within AR that in addition to blocking coactivator interactions attenuates the DNA binding of the receptor [58]. A phase II trial of Enza in AR+ TNBC is currently underway and initial results are favorable [14]. Traina et al. report a 16-week CBR of 35 %. The median PFS was 14.7 weeks and Enza was well-tolerated. An androgen-related gene signature was associated with a favorable clinical outcome. While a treatment devoid of chemotherapy side effects is intensely intriguing for patients and their providers, it is important to recognize that clinical investigators select patients with a more indolent clinical course to participate in these clinical trials, thus potentially enriching the patient population for those with more AR-driven tumors. Indeed, 55 % of the patients in this trial had >10 % AR expression.
Although not exclusively enrolling TNBC patients, other AR-directed therapy trials are underway including the CYP17 inhibitor abiraterone (NCT00755885), the androgens DHEA and CR1447 (4-OH-testosterone, NCT0200375, and NCT02067741, respectively), and antisense oligonucleotides targeting AR (NCT02144051). Multiple new generation AR inhibitors are also being tested in clinical trials for prostate cancer including ARN509 (NCT01946204) [59] and ODM-201 (NCT01429064) [60] that inhibit nuclear translocation. Until relatively recently, the discovery of AR antagonists was accomplished using empirical screens to identify molecules that exhibited high affinity receptor binding, the agonist/antagonist efficacy of which was subsequently determined using appropriate cellular/animal models. However, more contemporary, mechanism-based approaches have been developed that leverage the observation that the pharmacology of an AR ligand is determined by its impact on receptor structure and coregulator recruitment [58]. Using this approach, Joseph et al. identified a series of non-competitive AR antagonists which freeze AR in an “apo-conformation” and which attenuate AR action in models of castrate-resistant prostate cancer [61]. This general approach has also yielded AR antagonists which, by virtue of the conformational change they induce in AR, inhibit the activity of all of the clinically relevant AR mutations including F876L. For example, selective androgen receptor degraders (SARDs), compounds that induce proteasome dependent AR degradation and thus remove the receptor as a target for androgens and of pathways that converge on the receptor to enable ligand independent activation, have also been identified (D P McDonnell, JD Norris and J. Katzenellenbogen pers commun). An equally exciting new class of AR degraders was recently reported by Gustafson et al. where hydrophobic moieties (degrons) are attached to a high affinity AR ligand. The degron functionality targets the occupied receptor to the proteasome where it is quantitatively degraded [62]. The evaluation of several of these functionally distinct antagonists in breast cancer is currently underway.
Discussion
TNBC is an aggressive BC subtype for which no effective targeted therapies are available. While the exact percentages vary across studies, it is clear that a significant percentage of TNBCs express AR. Nuclear localization of the receptor suggests active AR signaling, and preclinical data indicates that AR + TNBC may critically depend on AR signaling for growth. The existence of the LAR TNBC subtype, with strong AR expression driving a luminal-like expression pattern in the absence of ER, is evidence that AR signaling can play a strong role in the biology of TNBC tumors. AR expression is associated with decreased proliferation in TNBC, but LAR tumors have a particularly poor prognosis, possibly because of their poor response to chemotherapy. Anti-androgens have shown particular efficacy in preclinical studies of LAR models and may be useful in improving the treatment of LAR tumors. Importantly, response to AR antagonists may not be limited to the LAR subtype, as the results of preclinical studies performed in vitro and in vivo have demonstrated that some cell lines with relatively low AR expression are sensitive to the newer generation anti-androgen Enza.
Recently reported and ongoing clinical trials using bicalutamide or Enza in TNBC have shown an increase in PFS, suggesting that AR-targeted therapies may improve patient prognosis and supporting a reclassification of TNBC into AR+ and “quadruple negative” disease. To date, there have been many classifications of TNBC subtypes which will likely lead to novel targeted therapeutics. However, we propose prioritizing the classification of AR+ and AR− disease because, unlike the other TNBC subtypes, the therapeutic target is clear, FDA-approved AR-targeted therapies are available, and early clinical trials demonstrate patient benefit from treatment with AR antagonists. Endocrine-targeted therapies such as tamoxifen and aromatase inhibitors have greatly improved the outcomes of ER+ breast cancer, and AR-targeted therapies have improved the prognosis of prostate cancer. Thus, treatment of hormone-dependent cancers has significantly benefited from endocrine-targeted therapy. Although profiling has identified multiple TNBC subtypes, it is reasonable to prioritize classification of AR+ and AR− disease as it is most likely to improve patient outcomes in the near future.
Additional research is needed to identify AR+ TNBC patients who will respond to AR-targeted therapies. Indeed, a signature of genes may more reliably predict responsiveness to anti-androgens than levels of AR alone and such a signature will likely be forthcoming from current trials of Enza in AR + TNBC. Furthermore, preclinical studies are needed to determine if AR-targeted therapies will be most effective if administered concurrently with chemotherapy, following chemotherapy, or perhaps as a targeted alternative to chemotherapy in AR+ TNBC patients with tumors expressing an “AR-responsive signature.” Further preclinical modeling will also determine whether anti-androgen therapy might synergize with other targeted therapies of current interest such as mTOR, CDK4/6, or EGFR inhibitors. It is also possible that a subset of AR+/HER2 overexpressing TNBC might be sensitive to the combination of an anti-androgen with HER2-directed therapy. Finally, research to determine possible mechanisms of resistance to anti-androgen therapy in TNBC cell lines by methods such as synthetic lethal screening and gene expression profiling of tumor specimens from the Enza trial and others will help to inform future clinical trial design and improve therapy for AR+ TNBC patients. The discovery that a hormone receptor with multiple FDA-approved antagonists may be critical for growth of a subset of TNBCs is an exciting development. Further preclinical research with AR-targeting drugs as single agents, combined with chemotherapy, or rationally determined targeted therapies, and then ultimately further clinical trials will establish whether an AR-directed agent will represent the first effective targeted therapy for AR+ TNBC.
References
Brenton JD, Carey LA, Ahmed AA, Caldas C (2005) Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin Oncol 23(29):7350–7360. doi:10.1200/JCO.2005.03.3845
Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA (2007) Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13(15 Pt 1):4429–4434. doi:10.1158/1078-0432.CCR-06-3045
Barton VN, D'Amato NC, Gordon MA, Lind HT, Spoelstra NS, Babbs BL, Heinz RE et al (2015) Multiple molecular subtypes of triple-negative breast cancer critically rely on androgen receptor and respond to enzalutamide in vivo. Mol Cancer Ther. doi:10.1158/1535-7163.MCT-14-0926
Micello D, Marando A, Sahnane N, Riva C, Capella C, Sessa F (2010) Androgen receptor is frequently expressed in HER2-positive, ER/PR-negative breast cancers. Virchows Arch 457(4):467–476. doi:10.1007/s00428-010-0964-y
Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM (2011) Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod Pathol 24(7):924–931. doi:10.1038/modpathol.2011.54
Mrklic I, Pogorelic Z, Capkun V, Tomic S (2013) Expression of androgen receptors in triple negative breast carcinomas. Acta Histochem 115(4):344–348. doi:10.1016/j.acthis.2012.09.006
Safarpour D, Pakneshan S, Tavassoli FA (2014) Androgen receptor (AR) expression in 400 breast carcinomas: is routine AR assessment justified? Am J Cancer Res 4(4):353–368
Thike AA, Yong-Zheng Chong L, Cheok PY, Li HH, Wai-Cheong Yip G, Huat Bay B, Tse GM, Iqbal J, Tan PH (2014) Loss of androgen receptor expression predicts early recurrence in triple-negative and basal-like breast cancer. Mod Pathol 27(3):352–360. doi:10.1038/modpathol.2013.145
Qi JP, Yang YL, Zhu H, Wang J, Jia Y, Liu N, Song YJ et al (2012) Expression of the androgen receptor and its correlation with molecular subtypes in 980 chinese breast cancer patients. Breast Cancer (Auckl) 6:1–8. doi:10.4137/BCBCR.S8323
Gucalp A, Tolaney S, Isakoff SJ, Ingle JN, Liu MC, Carey LA, Blackwell K et al (2013) Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin Cancer Res 19(19):5505–5512. doi:10.1158/1078-0432.CCR-12-3327
Traina, T. A. 2014. Stage I results from MDV3100-11: a 2 stage study of enzalutamide (Enza), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). Paper presented at the 37th annual San Antonio Breast Cancer Symposium
McNamara KM, Yoda T, Miki Y, Nakamura Y, Suzuki T, Nemoto N, Miyashita M et al (2014) Androgen receptor and enzymes in lymph node metastasis and cancer reoccurrence in triple-negative breast cancer. Int J Biol Markers. doi:10.5301/jbm.5000132
Cimino-Mathews A, Hicks JL, Illei PB, Halushka MK, Fetting JH, De Marzo AM, Park BH, Argani P (2012) Androgen receptor expression is usually maintained in initial surgically resected breast cancer metastases but is often lost in end-stage metastases found at autopsy. Hum Pathol 43(7):1003–1011. doi:10.1016/j.humpath.2011.08.007
Traina TA, Miller K, Yardley DA, O'Shaughnessy J, Cortes J, Awada A, Kelly CM, Trudeau ME, Schmid P, Gianni L, Garcia-Estevez L, Nanda R, Ademuyiwa FO, Chan S, Steinberg JL, Blaney ME, Tudor IC, Uppal H, Peterson AC, Hudis CA (2015) Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). Paper presented at the American Society of Clinical Oncology, Chicago, Illinois
Vera-Badillo FE, Templeton AJ, de Gouveia P, Diaz-Padilla I, Bedard PL, Al-Mubarak M, Seruga B, Tannock IF, Ocana A, Amir E (2014) Androgen receptor expression and outcomes in early breast cancer: a systematic review and meta-analysis. J Natl Cancer Inst 106(1):djt319. doi:10.1093/jnci/djt319
Anestis A, Karamouzis MV, Dalagiorgou G, Papavassiliou AG (2015) Is androgen receptor targeting an emerging treatment strategy for triple negative breast cancer? Cancer Treat Rev 41(6):547–553. doi:10.1016/j.ctrv.2015.04.009
Hickey TE, Robinson JL, Carroll JS, Tilley WD (2012) Minireview: the androgen receptor in breast tissues: growth inhibitor, tumor suppressor, oncogene? Mol Endocrinol 26(8):1252–1267. doi:10.1210/me.2012-1107
Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez-Angulo AM, Meric-Bernstam F, Valero V et al (2013) Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res 19(19):5533–5540. doi:10.1158/1078-0432.CCR-13-0799
Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. 2005. Lancet 365 (9472):1687–1717. doi:10.1016/S0140-6736(05)66544-0
Gradishar WJ (2010) Adjuvant endocrine therapy for early breast cancer: the story so far. Cancer Investig 28(4):433–442. doi:10.3109/07357901003631098
Gibson L, Lawrence D, Dawson C, Bliss J (2009) Aromatase inhibitors for treatment of advanced breast cancer in postmenopausal women. Cochrane Database Syst Rev 4, CD003370. doi:10.1002/14651858.CD003370.pub3
Robertson JF, Llombart-Cussac A, Rolski J, Feltl D, Dewar J, Macpherson E, Lindemann J, Ellis MJ (2009) Activity of fulvestrant 500 mg versus anastrozole 1 mg as first-line treatment for advanced breast cancer: results from the FIRST study. J Clin Oncol 27(27):4530–4535. doi:10.1200/JCO.2008.21.1136
Proctor E, Kidwell KM, Jiagge E, Bensenhaver J, Awuah B, Gyan K, Toy K et al (2015) Characterizing breast cancer in a population with increased prevalence of triple-negative breast cancer: androgen receptor and ALDH1 expression in Ghanaian women. Ann Surg Oncol. doi:10.1245/s10434-015-4455-x
McNamara KM, Yoda T, Nurani AM, Shibahara Y, Miki Y, Wang L, Nakamura Y et al (2014) Androgenic pathways in the progression of triple-negative breast carcinoma: a comparison between aggressive and non-aggressive subtypes. Breast Cancer Res Treat 145(2):281–293. doi:10.1007/s10549-014-2942-6
Doberstein K, Milde-Langosch K, Bretz NP, Schirmer U, Harari A, Witzel I, Ben-Arie A et al (2014) L1CAM is expressed in triple-negative breast cancers and is inversely correlated with androgen receptor. BMC Cancer 14:958. doi:10.1186/1471-2407-14-958
Abd-Elazeem MA (2015) Claudin 4 expression in triple-negative breast cancer: correlation with androgen receptors and Ki-67 expression. Ann Diagn Pathol 19(1):37–42. doi:10.1016/j.anndiagpath.2014.10.003
Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, Konishi H et al (2012) The growth response to androgen receptor signaling in ER alpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res 14(1):R27. doi:10.1186/bcr3112
Zhu X, Daffada AA, Chan CM, Dowsett M (1997) Identification of an exon 3 deletion splice variant androgen receptor mRNA in human breast cancer. Int J Cancer 72(4):574–580
Hu DG, Hickey TE, Irvine C, Wijayakumara DD, Lu L, Tilley WD, Selth LA, Mackenzie PI (2014) Identification of androgen receptor splice variant transcripts in breast cancer cell lines and human tissues. Horm Cancer 5(2):61–71. doi:10.1007/s12672-014-0171-4
Ahrens-Fath I, Politz O, Geserick C, Haendler B (2005) Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J 272(1):74–84. doi:10.1111/j.1742-4658.2004.04395.x
Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Kong X et al (2009) A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res 69(6):2305–2313. doi:10.1158/0008-5472.CAN-08-3795
Guedj M, Marisa L, de Reynies A, Orsetti B, Schiappa R, Bibeau F, MacGrogan G et al (2012) A refined molecular taxonomy of breast cancer. Oncogene 31(9):1196–1206. doi:10.1038/onc.2011.301
Doane AS, Danso M, Lal P, Donaton M, Zhang L, Hudis C, Gerald WL (2006) An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 25(28):3994–4008. doi:10.1038/sj.onc.1209415
Farmer P, Bonnefoi H, Becette V, Tubiana-Hulin M, Fumoleau P, Larsimont D, Macgrogan G et al (2005) Identification of molecular apocrine breast tumours by microarray analysis. Oncogene 24(29):4660–4671. doi:10.1038/sj.onc.1208561
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121(7):2750–2767. doi:10.1172/JCI45014
Burstein MD, Tsimelzon A, Poage GM, Covington KR, Contreras A, Fuqua SA, Savage MI et al (2015) Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res 21(7):1688–1698. doi:10.1158/1078-0432.CCR-14-0432
Jezequel P, Loussouarn D, Guerin-Charbonnel C, Campion L, Vanier A, Gouraud W, Lasla H et al (2015) Gene-expression molecular subtyping of triple-negative breast cancer tumours: importance of immune response. Breast Cancer Res 17(1):43. doi:10.1186/s13058-015-0550-y
Kemppainen JA, Wilson EM (1996) Agonist and antagonist activities of hydroxyflutamide and Casodex relate to androgen receptor stabilization. Urology 48(1):157–163
Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R et al (2012) Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 367(13):1187–1197. doi:10.1056/NEJMoa1207506
Lehmann BD, Bauer JA, Schafer JM, Pendleton CS, Tang L, Johnson KC, Chen X et al (2014) PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res 16(4):406. doi:10.1186/s13058-014-0406-x
Robinson JL, Macarthur S, Ross-Innes CS, Tilley WD, Neal DE, Mills IG, Carroll JS (2011) Androgen receptor driven transcription in molecular apocrine breast cancer is mediated by FoxA1. EMBO J 30(15):3019–3027. doi:10.1038/emboj.2011.216
Cochrane DR, Bernales S, Jacobsen BM, Cittelly DM, Howe EN, D'Amato NC, Spoelstra NS et al (2014) Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res 16(1):R7. doi:10.1186/bcr3599
Ni M, Chen Y, Lim E, Wimberly H, Bailey ST, Imai Y, Rimm DL, Liu XS, Brown M (2011) Targeting androgen receptor in estrogen receptor-negative breast cancer. Cancer Cell 20(1):119–131. doi:10.1016/j.ccr.2011.05.026
Vranic S, Gatalica Z, Wang ZY (2011) Update on the molecular profile of the MDA-MB-453 cell line as a model for apocrine breast carcinoma studies. Oncol Lett 2(6):1131–1137. doi:10.3892/ol.2011.375
Bartlett JM, Ibrahim M, Jasani B, Morgan JM, Ellis I, Kay E, Connolly Y et al (2009) External quality assurance of HER2 FISH and ISH testing: three years of the UK national external quality assurance scheme. Am J Clin Pathol 131(1):106–111. doi:10.1309/AJCPLN78ZQXEMNMA
Okita R, Mougiakakos D, Ando T, Mao Y, Sarhan D, Wennerberg E, Seliger B, Lundqvist A, Mimura K, Kiessling R (2012) HER2/HER3 signaling regulates NK cell-mediated cytotoxicity via MHC class I chain-related molecule A and B expression in human breast cancer cell lines. J Immunol 188(5):2136–2145. doi:10.4049/jimmunol.1102237
Bartlett JM, Ibrahim M, Jasani B, Morgan JM, Ellis I, Kay E, Magee H, Barnett S, Miller K (2007) External quality assurance of HER2 fluorescence in situ hybridisation testing: results of a UK NEQAS pilot scheme. J Clin Pathol 60(7):816–819. doi:10.1136/jcp.2006.040840
Wolff AC, Hammond ME, Hicks DG, Dowsett M, McShane LM, Allison KH, Allred DC et al (2013) Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol 31(31):3997–4013. doi:10.1200/JCO.2013.50.9984
Narayan M, Wilken JA, Harris LN, Baron AT, Kimbler KD, Maihle NJ (2009) Trastuzumab-induced HER reprogramming in "resistant" breast carcinoma cells. Cancer Res 69(6):2191–2194. doi:10.1158/0008-5472.CAN-08-1056
O'Brien NA, Browne BC, Chow L, Wang Y, Ginther C, Arboleda J, Duffy MJ, Crown J, O'Donovan N, Slamon DJ (2010) Activated phosphoinositide 3-kinase/AKT signaling confers resistance to trastuzumab but not lapatinib. Mol Cancer Ther 9(6):1489–1502. doi:10.1158/1535-7163.MCT-09-1171
Le Du F, Eckhardt BL, Lim B, Litton JK, Moulder S, Meric-Bernstam F, Gonzalez-Angulo AM, Ueno NT (2015) Is the future of personalized therapy in triple-negative breast cancer based on molecular subtype? Oncotarget 6(15):12890–12908
Moore NL, Buchanan G, Harris JM, Selth LA, Bianco-Miotto T, Hanson AR, Birrell SN, Butler LM, Hickey TE, Tilley WD (2012) An androgen receptor mutation in the MDA-MB-453 cell line model of molecular apocrine breast cancer compromises receptor activity. Endocr Relat Cancer 19(4):599–613. doi:10.1530/ERC-12-0065
Cuenca-Lopez MD, Montero JC, Morales JC, Prat A, Pandiella A, Ocana A (2014) Phospho-kinase profile of triple negative breast cancer and androgen receptor signaling. BMC Cancer 14:302. doi:10.1186/1471-2407-14-302
Traina, TA, Gucalp A, Polkinghorn W, Isakoff S, Tolaney S, Carey L, Ingle J, Nabell L, Forero A, Rugo H, Blackwell K, Liu M, Soloway M, Mose L, Giri D, Viale A, Hudis C, Sawyers C, Parker J. 2014. Whole transcriptome analysis of AR+ ER/PR- metastatic breast cancers treated with bicalutamide on TBCRC011. Paper presented at the San Antonio Breast Cancer Symposium
Cuenca-Lopez MD, Andrades JA, Gomez S, Zamora-Navas P, Guerado E, Rubio N, Blanco J, Becerra J (2014) Evaluation of posterolateral lumbar fusion in sheep using mineral scaffolds seeded with cultured bone marrow cells. Int J Mol Sci 15(12):23359–23376. doi:10.3390/ijms151223359
Graham TR, Yacoub R, Taliaferro-Smith L, Osunkoya AO, Odero-Marah VA, Liu T, Kimbro KS, Sharma D, O'Regan RM (2010) Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res Treat 123(1):139–147. doi:10.1007/s10549-009-0623-7
Osguthorpe DJ, Hagler AT (2011) Mechanism of androgen receptor antagonism by bicalutamide in the treatment of prostate cancer. Biochemistry 50(19):4105–4113. doi:10.1021/bi102059z
Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, Cui H et al (2009) Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol 16(4):452–460. doi:10.1016/j.chembiol.2009.01.016
Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, Chen Y et al (2012) ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res 72(6):1494–1503. doi:10.1158/0008-5472.CAN-11-3948
ODM-201 is safe and active in metastatic castration-resistant prostate cancer. 2014. Cancer Discov 4 (9):OF10. doi:10.1158/2159-8290.CD-RW2014-150
Joseph JD, Wittmann BM, Dwyer MA, Cui H, Dye DA, McDonnell DP, Norris JD (2009) Inhibition of prostate cancer cell growth by second-site androgen receptor antagonists. Proc Natl Acad Sci U S A 106(29):12178–12183. doi:10.1073/pnas.0900185106
Gustafson JL, Neklesa TK, Cox CS, Roth AG, Buckley DL, Tae HS, Sundber TB, Stagg DB, Hines J, McDonnell DP, Norris JD, Crews CM. 2015. Small molecule mediated degradation of the androgen receptor through hydrophobic tagging. Angewandte Chemie
Conflict of Interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Barton, V.N., D’Amato, N.C., Gordon, M.A. et al. Androgen Receptor Biology in Triple Negative Breast Cancer: a Case for Classification as AR+ or Quadruple Negative Disease. HORM CANC 6, 206–213 (2015). https://doi.org/10.1007/s12672-015-0232-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-015-0232-3