
Abstract
Therapy-resistant cancer cells are a major problem in cancer research. Recent studies suggest that the epithelial-mesenchymal transition (EMT) is a key mechanism in therapy resistance. Yet, the expressions of EMT markers, EMT core regulators, and a stem cell marker of BMI1 during chemotherapy have been poorly analyzed in clinical breast cancer specimens. In the present study, we investigated the roles of RhoC under chemotherapy to follow up on earlier findings demonstrating the involvement of RhoC in prostate cancer resistance to endocrine therapy. Immunohistochemically, E-cadherin expression was significantly lower in human breast cancer specimens analyzed after chemotherapy than specimens biopsied before chemotherapy. Significant upregulation of fibronectin, a mesenchymal EMT marker, was found in post-chemotherapy analysis. A study of the EMT core regulators of SNAIL1, SNAIL2, TWIST1, and a well-known stem cell marker of BMI1 revealed no post-chemotherapy upregulation of these molecules. In contrast, RhoC expression was significantly upregulated in post-chemotherapy breast cancer specimens. MCF-7 cells stably transfected with the constitutive active (CA) RhoC plasmid manifested a reduced level of E-cadherin at the peripheries and disorganization of actin fibers, with no accompanying upregulation of SNAIL1, SNAIL2, TWIST1, or BMI1 in Western blots. Exposure of etoposide on MCF-7 cells showed RhoC upregulation together with reduced membranous expression of E-cadherin and disorganization of actin fibers. In MTT assay, however, the CA-RhoC-expressing MCF-7 cells failed to show chemotherapy resistance under etoposide treatment. Taken in sum, RhoC may contribute to an EMT-like process in human breast cancer during chemotherapy.
Similar content being viewed by others

Introduction
Breast cancer is the leading cause of cancer death in women worldwide. Recent advances in molecular biology allow us to draw a clear landscape of the molecular networks in breast cancer [1]. Highly effective drugs are being continuously developed based on this information [2]. Yet, novel therapeutic approaches are still clearly needed, as tumors tend to relapse after undergoing existing therapies of various types. Recent studies have revealed that the epithelial-mesenchymal transition (EMT) takes part in chemotherapy and endocrine therapy resistance in breast cancer [3–5]. The EMT is also shown to be involved in the generation of cancer stem cells (CSCs) [6], a class of cells believed to be highly therapy resistant [7]. Further, the CSC-related molecule BMI1 has been shown to both contribute to chemotherapy resistance [8] and induce the EMT [9]. These findings, taken together, suggest that the EMT and CSCs are strongly linked to chemotherapy resistance. The EMT was originally described as a loss of E-cadherin and gain of mesenchymal markers in cancer cells, a process that mimicked mesoderm and neural crest formation during embryogenesis [10, 11]. The Snail and Twist homologs are defined as core transcriptional repressors that directly bind to the E-cadherin promoter [10, 11]. While several immunohistochemical analyses have focused on the expressions of BMI1 and the Snail and Twist homologs in human breast cancer [12, 13], none before now have compared these molecules in human breast cancer specimens of the same patient before and after chemotherapy.
We have recently shown that RhoC is linked to cancer cell survival through a partial epithelial-mesenchymal transition (EMT) process after endocrine therapy in cultured mouse breast cancer cells and human prostate cancer specimens [14]. The Rho small GTPases are major regulators of actin dynamics and play a role in cell motility [15]. The Rho GTPases are subdivided into three subfamilies respectively descended from the prototypal proteins RhoA, Rac1, and Cdc42 [16]. The activation of RhoA, RhoB, and RhoC, the three highly homologous isoforms of the RhoA family, facilitates amoeboid cell movement by stimulating the organization of actin stress fibers (ASFs) [15, 16]. Among the three isoforms, RhoC was found to play a key role in tumor progression [17–20]. Two earlier studies on human breast cancer have shown a correlation between RhoC expression and aggressive phenotypes [21, 22]. Little is known, however, about the roles of RhoC in human breast cancer during chemotherapy.
In the present comparison of human breast cancer specimens between pre- and post-chemotherapy, the upregulation of RhoC and the downregulation of E-cadherin were both significantly increased after the chemotherapy. In contrast, we found no significant upregulation of SNAIL1, SNAIL2, TWIST1, or BMI1. Lastly, transfection of the constitutive active (CA) RhoC plasmid into human breast cancer MCF-7 cells reduced the membranous localization of E-cadherin. These findings implicate RhoC as a key factor in breast cancer progression under chemotherapy.
Materials and Methods
Cases
Tumor specimens from 50 breast cancer cases resected after chemotherapy were retrieved from the archives of the Pathology Department of Jichi Medical University Hospital. All cases, diagnosed as invasive ductal carcinomas, had undergone core needle biopsy before chemotherapy, and each biopsied specimen was compared to the resected specimen from the same patient. The patient characteristics are summarized in Table 1 and shown in detail in Supplementary Table S1. The chemotherapy protocols, which varied considerably from case to case, are also shown in Supplementary Table S1. Data on the estrogen receptor (ER), progesterone receptor (PgR), and Her2 were retrieved from the clinical records. The duration of chemotherapy was 63 to 504 days (mean 152 days). Immunohistochemical studies were performed with the approval of the local ethics committee at Jichi Medical University.
Cells
Estrogen-sensitive human breast cancer MCF-7 cells were maintained in a DMEM medium supplemented with 10 % fetal bovine serum (FBS) in the presence of insulin (4 μg/ml) and 17β-estradiol (E2) (100 nM).
Plasmids, Mutagenesis, and Isolation of Stable Transfectant Cells
The method for constructing the mouse constitutive active (CA) RhoC (G12V) expression plasmid was described previously [14]. MCF-7 cells were transfected with the CA-RhoC plasmid by a Lipofectamin (Invitrogen, Carlsbad, CA) method and then selected using 800 μg/ml of G418 sulfate (GIBCO/Invitrogen). Two clones with standard expression of the tagged protein in Western blots were used in further experiments.
Immunohistochemistry and Antibodies
Supplementary Table S2 summarizes the antibodies used in this study and the methods for antigen retrieval and detection. In general, formalin-fixed and paraffin-embedded sections were pre-treated in a microwave oven or an autoclave for the indicated times. Next, the sections were allowed to interact with each antibody at 4 °C overnight, subjected to either standard treatment with avidin-biotin complex or incubation with Envision (DAKO JAPAN, Tokyo, Japan), and stained with DAB. The immunostaining was scored as follows according to the criteria previously reported by our group [14]: (−), negative; (±), weakly positive in less than 20 % of cancer cells; (+), distinctly positive in 20 to 80 % of cancer cells; and (++), strongly positive in more than 80 % of cancer cells.
Rho Activation Assay
An Rho activation assay was performed using the Rho Activation Assay Kit (Upstate/MILLIPORE, Temecula, CA) by the method described in our earlier report [14]. Briefly, cell extracts were prepared by incubation for 48 h in a DMEM medium containing 10 % FBS in the presence of E2 and insulin. The extracted proteins were incubated with Rhotekin Rho-binding domain agarose beads for 45 min at 4 °C with gentle agitation. The beads were washed three times with the buffer provided, and then the pellets were resolved by a standard sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to the polyvinylidene difluoride (PVDF) membrane. The blots were incubated with the Rho antibody provided in the kit and then detected with an ECL prime kit (GE Healthcare, Buckinghamshire, UK).
Western Blots
Cell extracts were prepared in the DMEM-maintained medium in a sub-confluent condition with a cell lysis reagent of CelLytic M (Sigma-Aldrich, St. Louis, MO). In the study of RhoC expression under etoposide treatment, MCF-7 cells (1.2 × 106/dish) were plated in a phenol-red-free RPMI 1640 medium supplemented with 2 % FBS treated with dextran-coated charcoal (dcc) in the absence of E2 and insulin. The cells were further cultured for 5 days in a phenol-red-free RPMI 1640 medium containing 2 % dcc-treated FBS in the absence or presence of 100 nM E2 and/or 10 μM of etoposide (Wako Pure Chemical Industries, Osaka, Japan), with medium changes every other days. Cell extracts were similarly prepared. In general, 20 μg of lysates was separated by SDS-PAGE electrophoresis and transferred onto PVDF membranes. The blots were reacted with the antibodies listed in Supplementary Table S2, and the signals were detected with an ECL prime kit. For loading controls, the membranes were stripped with reprobe buffer [62.5 mM Tris–HCl (pH 6.8), 100 mM 2-mercaptoethanol, 2 % SDS] at 60 °C for 30 min and then immunoblotted with an anti-GAPDH antibody (MILLIPORE, Temecula, CA) followed by the secondary antibody.
Phalloidin Staining and Immunocytochemistry of E-cadherin
The method for phalloidin staining was described previously [14]. Briefly, MCF-7 cells (1.2 × 104/well) were plated onto a 4-well chamber slide. The next day, the medium was changed to the DMEM-maintained medium. After a 6 h culture, the cells were fixed in a PBS solution with 4 % paraformaldehyde for 60 min, treated with 0.1 % Triton-X for 5 min, and stained with rhodamine-labeled phalloidin. The specimens were observed with a fluorescence microscope. In the study of etoposide treatment, cells were plated and culture onto a 4-well chamber slide using a phenol-red-free RPMI1640 medium containing 2 % dcc FBS in the absence or presence of E2 and/or etoposide. After culture for 5 days, cells were similarly fixed and stained with rhodamine-labeled phalloidin. In immunocytochemistry of E-cadherin, cells were similarly cultured, fixed, incubated with the anti-E-cadherin antibody, and then reacted with a fluorescein isothiocyanate (FITC)-conjugated secondary antibody. The slide was observed with a fluorescence microsocpe.
MTT Assay
Parental and transfectant MCF-7 cells (6.0 × 103/well) were plated onto a 96-well plate in a phenol-red-free RPMI 1640 medium supplemented with 2 % dcc-treated FBS in the absence of insulin and E2. The next day (day 1), the medium was changed to the phenol-red-free RPMI 1640 medium with 2 % dcc-treated FBS in the presence or absence of E2 and/or 10 μM of etoposide. The medium was changed once on day 3, and MTT was added to the medium at a final concentration of 1 mg/ml on day 5. After 4 h of incubation with the MTT, formazan substrates were collected, resolved with DMSO, and measured at an absorbance of 570 nm, with reference at 690 nm.
Statistical Analysis
The statistical significance of value changes was determined by the Wilcoxon signed-ranks test or Mann-Whitney’s U test using Statcel software (OMS, Saitama, Japan). Results were considered significant when the P values were less than 0.05.
Results
Membranous E-cadherin localization is reduced after chemotherapy in human breast cancer
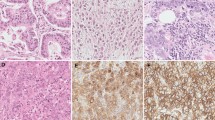
To confirm the involvement of the EMT process in chemotherapy resistance, we began our experiment by comparing E-cadherin expression in cancer specimens biopsied before surgery with specimens surgically resected after chemotherapy. In spite of considerable variation in the chemotherapy protocols from case to case, E-cadherin expression was significantly more downregulated after chemotherapy compared to before (P < 0.0002) (Fig. 1a, b and Table 2). Next, we sought to determine whether any enhancement of the nuclear translocation of β-catenin accompanied the reduction of E-cadherin. β-catenin expression in cytoplasmic membranes was unchanged after chemotherapy (P = 0.248), and the nuclear translocation of β-catenin was almost undetectable in all but one post-chemotherapy specimen (Fig. 1c, d and Table 2). On examination of mesenchymal markers, significant upregulation was found in fibronectin, but not in N-cadherin, between pre- and post-chemotherapy (P < 0.0001 and P = 0.317) (Fig. 1e–h and Table 2). The induction of a partial EMT seems very likely after breast cancer chemotherapy, given the great importance of the membranous expression of E-cadherin as a factor in epithelial phenotypes.

Immunohistochemical comparison of E-cadherin, β-catenin, N-cadherin, and fibronectin between pre- and post-chemotherapy of the same case in human breast carcinoma specimens. Immunostaining of E-cadherin (a, b), β-catenin (c, d), N-cadherin (e, f), and fibronectin (g, h) in human breast cancer specimens surgically resected after chemotherapy (after) (a, c, e, g) and in biopsied specimen of the same case before chemotherapy (before) (b, d, f, h). The arrowhead indicates nuclear translocation of β-catenin in one case (d). The immunohistochemical scoring is also shown as (−), (±), (+), and (++)
Neither SNAIL1, SNAIL2, TWIST1, nor BMI1 expression was upregulated after chemotherapy in human breast cancer.
Next, we compared the expressions of SNAIL1, SNAIL2, TWIST1, and BMI1 between pre- and post-chemotherapy. The nuclear expressions of SNAIL1, SNAIL2, and TWIST1 were shown to be significantly downregulated, not upregulated, after chemotherapy (P < 0.05) (Fig. 2a–f and Table 3). We also observed a significant downregulation of BMI1 expression after chemotherapy (P < 0.05) (Fig. 2g, h and Table 3). These findings suggest that SNAIL1, SNAIL2, TWIST1, and BMI1 are less likely to be involved in the partial EMT under chemotherapy in human breast cancer.

Immunohistochemical comparison of SNAIL1, SNAIL2, TWIST1, and BMI1 between pre- and post-chemotherapy of the same case in human breast carcinoma specimens. Immunostaining of SNAIL1 (a, b), SNAIL2 (c, d), TWIST1 (e, f), and BMI1 (g, h) in human breast cancer specimens surgically resected after chemotherapy (after) (a, c, e, g) and in biopsied specimen of the same case before chemotherapy (before) (b, d, f, h). The immunohistochemical scoring is also shown
RhoC is expressed at a higher level after chemotherapy than before in human breast cancer.
Rho small GTPases are well established to play a key role in cytoskeletal actin dynamics involved in the regulation of cadherin-mediated cell-cell adhesion. Our earlier data on the involvement of RhoC in endocrine therapy resistance in prostate cancer [14] prompted us to compare the expressions of RhoC and NET1 (a Rho-specific guanine exchange factor) in human breast cancer specimens between pre- and post-chemotherapy. RhoC expression was significantly more upregulated in the specimens resected after chemotherapy than in the specimens biopsied before the therapy (P < 0.0002) (Fig. 3a, b and Table 3). In contrast to our earlier finding in prostate cancer [14], the expression of NET1 was significantly downregulated after the therapy (P < 0.0001) (Fig. 3c, d and Table 3). This alteration in RhoC expression suggests that other Rho guanine nucleotide exchange factors may be involved in RhoC activation in breast cancer. The findings also suggest that RhoC activation has the potential to reduce the membranous expression of E-cadherin, possibly by regulating actin dynamics, under human breast cancer chemotherapy.

Immunohistochemical comparison of RhoC and NET1 between pre- and post-chemotherapy in human breast carcinoma specimens. RhoC (a, b) and NET1 (c, d) immunostaining in a breast cancer specimen surgically resected after chemotherapy (after) (a, c) and in the biopsied specimen of the same case before chemotherapy (before) (b, d). The immunohistochemical scoring is also shown
Stable transfection with a CA-RhoC plasmid into MCF-7 cells reduces membranous E-cadherin expression and disrupts the organization of actin fibers.
We next investigated whether or not stable transfection of a CA-RhoC expression plasmid into MCF-7 cells induces an EMT process. An Rho activation assay (Fig. 4a) confirmed the function of the transfected gene by revealing higher Rho activity in MCF-7 cells expressing CA-RhoC than in parental MCF-7 cells. In immunocytochemistry and phalloidin staining, the CA-RhoC-expressing cells exhibited reduced E-cadherin expression at the peripheries of the cancer cell nests and disorganized actin fibers (Fig. 4c, d, f, g). In contrast, no signs of E-cadherin loss or actin fiber disorganization were observed in the parental MCF-7 cells (Fig. 4b, e). In Western blots using whole cell lysates, the overall E-cadherin levels were comparable in the transfectant and parental cells (Fig. 4h), suggesting that membranous E-cadherin expression is disorganized in the CA-RhoC-expressing cells. Nuclear localization of β-catenin was undetectable in CA-RhoC-expressing MCF-7 cells (data not shown). No significant upregulation of SNAIL1, SNAIL2, TWIST1, or BMI1 was found in CA-RhoC-expressing MCF-7 cells versus the parental cells in Western blots (Fig. 4h). These observations suggest that RhoC is one of the regulatory factors involved in the reduction of the membranous localization of E-cadherin.

Reduction of E-cadherin and disorganization of actin fibers in CA-RhoC-expressing MCF-7 cells. Rho activation assay in transfectant and parental MCF-7 cells (a). Cell extracts were prepared from subconfluent CA-RhoC-expressing MCF-7 cells and parental MCF-7 cells with the lysate solution in the kit. Four hundred micrograms of extracted protein was incubated with Rhotekin Rho-binding domain agarose beads. The pellets were analyzed by Western blots using the Rho antibody provided in the kit (Rho). The total levels of RhoA family GTPases (Total), namely, RhoA, RhoB, and RhoC, are shown at the bottom. Immunocytochemistry of E-cadherin (b, c, d). Transfectant (CA-RhoC-1, -2) and parental (MCF) MCF-7 cells (1.2 × 104/well) were plated onto a 4-well chamber slide. The medium was exchanged once on the next day and cultured for 6 h. The cells were fixed with 4 % paraformaldehyde, incubated with the anti-E-cadherin antibody, and reacted with an FITC-conjugated secondary antibody. The slide was observed with a fluorescence microscope. Phalloidin staining (e, f, g). Transfectant (CA-RhoC-1, -2) and parental (MCF) MCF-7 cells were similarly plated and cultured onto a 4-well chamber slide. The cells were fixed with 4 % paraformaldehyde, stained with rhodamine-labeled phalloidin, and observed with a fluorescence microscope. Western blots (h). Cell lysates were prepared from subconfluent CA-RhoC-expressing MCF-7 cells and the parental MCF-7 cells with the lysis solution indicated in the Materials and Methods section. The cell lysates (20 μg) were separated by SDS-PAGE electrophoresis and transferred onto PVDF membranes. The blots were reacted with the antibodies of E-cadherin, SNAIL1, SNAIL2, TWIST1, and BMI1. Signals were detected with an ECL prime kit. The bottom panel shows the loading controls of GAPDH
Rho activity is not directly linked with resistance to etoposide treatment in CA-RhoC-expressing MCF-7 cells.
We next analyzed effects of etoposide treatment on the EMT process and RhoC expression in MCF-7 cells. RhoC expression is elevated under etoposide treatment in the presence of E2 in MCF-7 cells (Fig. 5a). Although overall levels of E-cadherin expression were unchanged (Fig. 5a), immunocytochemistry displayed that membranous E-cadherin expression is reduced at the peripheries of cancer cell nests under etoposide treatment together with disorganization of actin fibers (Fig. 5b–i). Upon exposure to etoposide in the presence of E2, the cell viability of CA-RhoC-expressing MCF-7 cells was reduced to 49.8 and 51.4 %, levels similar to that observed in the parental cells (48.8 %) (Fig. 5j, k). In the absence of E2, the treatment with etoposide mildly lowered the cell viability to 94.2 % (P < 0.05) and 73.7 % (P = 0.275) in the transfectant cells, versus 67.9 % in the parental cells (Fig. 5j, k). In view of apoptosis, cleaved caspase 3, a well-known apoptotic marker, was faintly detected only in the parental MCF-7 cells in the presence of E2 (Supplementary Fig. S1). In TUNEL assay, labeled cells were at marginal levels in the parental and the trasfectant MCF-7 cells (data not shown). Both findings suggest that involvement of apoptosis is minimal under this dose of etoposide treatment. Taken in sum, RhoC may not directly contribute to chemotherapy resistance in MCF-7 cells.

Effects of etoposide treatment in MCF-7 cells. Western blots (a). MCF-7 cells (1.2 × 106/dish) were plated in a phenol-red-free RPMI 1640 medium supplemented with 2 % FBS treated with dextran-coated charcoal (dcc) in the absence of E2 and insulin. The cells were further cultured for 5 days in a phenol-red-free RPMI 1640 medium containing 2 % dcc-treated FBS in the absence or presence of 100 nM E2 and/or 10 μM of etoposide, with medium changes every other days. Cell extracts were prepared, and Western blots were similarly performed using anti-RhoC and anti-E-cadherin antibodies. The bottom panel shows the loading controls of GAPDH. Immunocytochemistry of E-cadherin (b, c, d, e) and phalloidin staining (f, g, h, i). MCF-7 cells were similarly plated and culture onto a 4-well chamber slide. After culture for 5 days, cells were fixed with 4 % paraformaldehyde. The chamber slides were either stained with rhodamine-labeled phalloidin or reacted with the anti-E-cadherin antibody and then with an FITC-conjugated secondary antibody. The slide was observed with a fluorescence microsocpe. MTT assay (j, k). Parental (MCF) and transfectant (CA-RhoC-1, -2) MCF-7 cells (6 × 103/well) were plated onto a 96-well plate in a phenol-red-free RPMI 1640 medium, supplemented with 2 % dcc-treated FBS. The next day, the cells were either left untreated or cultured with 10 μM etoposide (VP16) in the absence (−) or presence of 100 nM E2 (E2). The medium was changed every other day. MTT assays were performed after incubation for 4 more days. Percentages compared to the values of untreated cells are also shown. The other trial gave a similar result
Discussion
As long as we know, this is the first report to compare various EMT-related molecules in human breast cancer specimens of the same patient before and after chemotherapy. Through this comparison, we demonstrated a reduction of E-cadherin and an upregulation of fibronectin in clinical breast specimens resected after chemotherapy. In earlier studies, BMI1 and Snail and Twist homologs were found to take part in the EMT-mediated therapy resistance of tumor cells [23–28]. Unexpectedly, no evidence of SNAIL1, SNAIL2, TWIST1, or BMI1 upregulation was observed in the post-chemotherapy specimens examined in the present study. Interestingly, significantly more upregulation of RhoC was found in the specimens resected after chemotherapy versus the specimens biopsied before. Human breast cancer MCF-7 cells stably transfected with the CA-RhoC plasmid exhibited an EMT-like phenotype characterizing by reduced membranous E-cadherin expression and disorganization of actin fibers. In addition, RhoC was also upregulated in the similar EMT-like process under etoposide treatment in MCF-7 cells. Taken in sum, RhoC may be a key regulator in the EMT-like process in human breast cancer during chemotherapy. Notably, an earlier study showed that RhoA silencing reverts the resistance to doxorubicin in human colon cancer [29]. In another study, RhoC overexpression was found to be independently predictive of poor response to doxorubicin-based chemotherapy [22]. These findings tempt us to speculate that RhoC is involved in cell survival during chemotherapy in clinical breast cancer. The present study, however, failed to demonstrate significant chemotherapy resistance in CA-RhoC-expressing MCF-7 cells in MTT assay. It is likely that the involvement of the EMT-core regulators or BMI1 might be required for chemotherapy resistance through EMT. To explore this issue, we are now under investigation of cell survival signaling activated by EMT in clinical colon cancer specimens and cultured colon cancer cell lines.
In conclusion, the present study showed that RhoC is more upregulated in breast cancer specimens after chemotherapy than before. Further, CA-RhoC-expressing MCF-7 cells had the potential to reduce the membranous expression of E-cadherin. RhoC may be an attractive molecule to prevent the EMT process.
References
The Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490:61–70
Lee AV, Davidson NE (2013) Genomics, drug approval, and optimal treatment duration. Nat Rev Clin Oncol 11:71–72
Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R, Barrow D, Nicholson RI (2006) Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer 118:290–301
Creighton CJ, Li X, Landis M, Dixon M, Neumeister VM, Sjolund A, Rimm DL, Wong H, Rodriguez A, Herschkowitz JI, Fan C, Zhang X, He X, Pavlick A, Gutierrez MC, Renshaw L, Larionov AA, Faratian D, Hilsenbeck SG, Perou CM, Lewis MT, Rosen JM, Chang JC (2009) Residual breast cancers after conventional therapy display mesenchymal tumor-initiating features. Proc Natl Acad Sci U S A 106:13820–13825
Foroni C, Broggini M, Generali D, Damia G (2012) Epithelial-mesenchymal transition and breast cancer: role, molecular mechanisms and clinical impact. Cancer Treat Rev 38:689–697
Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Poylak K, Brisken C, Yang J, Weinberg RA (2008) The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133:704–715
Visvader JE, Lindeman GJ (2013) Cancer stem cells in solid tumors: accumulating evidence and unsolved questions. Nat Rev Cancer 8:755–768
Rahman H, Saleem M (2012) Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells 30:372–378
Song L-B, Li J, Liao W-T, Feng Y, Yu C-P, Hu L-J, Kong Q-L, Xu L-H, Zhang X, Liu W-L, Li M-Z, Zhang L, Kang T-B, Fu L-W, Huang W-L, Xia Y-F, Tsao SW, Li M, Band V, Shi Q-H, Zeng Y-X, Zeng M-S (2006) The polycomb group protein BMI1 represses the tumor suppressor PTEN and induces epithelial-mesenchymal transition in human nasopharyngeal epithelial cells. J Clin Invest 119:3626–3636
Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA (2000) The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol 2:76–83
Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia de Herreros A (2000) The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol 2:84–89
Joensuu K, Hagström J, Leidenius M, Haglund C, Andersson LC, Sariola H, Heikkilä P (2011) Bmi-1, c-myc, and Snail expression in primary breast cancers and their metastases-elevated Bmi1 expression in late breast cancer relapses. Virchow Arch 459:31–39
Van Nes JG, de Kruijf EM, Putter H, Faratian D, Munro A, Campbell F, Smit VT, Liefers GJ, Kuppen PJ, van de Velde CJ, Bartlett JM (2012) Co-expression of SNAIL and TWIST determines prognosis in estrogen receptor-positive early breast cancer patients. Breast Cancer Res Treat 133:49–59
Kawata H, Shimada N, Kamiakito T, Komatsu K, Morita T, Ota T, Obayashi M, Shitara K, Tanaka A (2012) RhoC and guanine nucleotide exchange factor Net1 in androgen-unresponsive mouse mammary carcinoma SC-4 cells and human prostate cancer after short-term endocrine therapy. Prostate 72:1071–1079
Olson EN, Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11:353–365
Heasman SJ, Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9:690–701
Clark EA, Golub TR, Lander ES, Hynes RO (2000) Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406:532–535
Shikada Y, Yoshino I, Okamoto T, Fukuyama S, Kameyama T, Maehara Y (2003) Higher expression of RhoC is related to invasiveness in non-small cell lung carcinoma. Clin Cancer Res 9:5282–5286
Wang W, Yang L-Y, Huang G-W, Lu W-Q, Yang Z-L, Yang J-Q, Liu H-L (2004) Genomic analysis reveals RhoC as a potent marker in hepatocellular carcinoma with poor prognosis. Br J Cancer 90:2349–2355
Hakem A, Sanchez-Sweatman O, You-Ten A, Duncan G, Wakeham A, Khokha H, Mak TW (2005) RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev 19:1974–1979
van Golen KL, Wu Z-F, Qiao XT, Bao LW, Merajver SD (2000) RhoC GTPase, a novel transforming oncogene for human mammary epithelial cells that partially recapitulates the inflammatory breast cancer phenotype. Cancer Res 60:5832–5838
Kleer CG, Griffith KA, Sabel MS, Gallagher G, van Golen KL, Wu Z-F, Merajver SD (2005) RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res Treat 93:101–110
Elloul S, Elstrand MB, Nesland JM, Tropé CG, Kvalheim G, Goldberg I, Reich R, Davidson B (2005) Snail, slug, and smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer 103:1631–1643
Moody SE, Perez D, Pan T-C, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA (2005) The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell 8:197–209
Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok J, Hartwell K, Richardson AL, Weinberg RA (2007) Mesenchyme forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci U S A 104:10069–10074
Zhang S, Balch C, Chan MW, Lai H-C, Matei D, Schilder JM, Yan PS, Huang TH-M, Nephew KP (2008) Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 68:4311–4320
Zhu Y, Yu F, Jiao Y, Feng J, Tang W, Yao H, Gong C, Chen J, Su F, Zhang Y, Song E (2011) Reduced miR-128 in breast tumor-initiating cells induces chemotherapeutic resistance via Bmi-1 and ABCC5. Clin Cancer Res 17:7105–7115
Kawamoto A, Yokoe T, Tanaka K, Saigusa S, Toiyama Y, Yasuda H, Inoue Y, Miki C, Kusunoki M (2012) Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol Rep 27:51–57
Doublier S, Riganti C, Voena C, Costamagna C, Aldieri E, Pescarmona G, Ghigo D, Bosia A (2008) RhoA silencing reverts the resistance to doxorubicin in human colon cancer cells. Mol Cancer Res 6:1607–1620
Acknowledgments
We are grateful to A. Fukushima, S. Itoh, M. Tamura, J. Kosegawa, M. Yamamoto, K. Honmou, S. Sato, M. Kikuchi, M. Hoshino, and Y. Haga at Jichi Medical University Hospital for their technical assistance. This study was supported in part by Grant-in-Aid of Young Scientists (B) of the Ministry of Education, Culture, Sports, Science and Technology (KAKENHI 26860244). This study was also supported in part by the Jichi Medical University Young Investigator Award.
Conflict of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary table 1
(XLS 30 kb)
Supplementary table 2
(XLS 21 kb)
Supplementary Fig. S1
by western blots in parental and CA-RhoC-expressing MCF-7 cells after 5 days culture in the absence or presence of E2 (E2) and/or VP-16 (VP16). The other CA-RhoC-expressing cell line gave a similar result. Anti-cleaved caspase-3 (Asp175) rabbit monoclonal antibody was purchased from Cell Siganaling Technology. (GIF 8 kb)
Rights and permissions
About this article

Cite this article
Kawata, H., Kamiakito, T., Omoto, Y. et al. RhoC Upregulation Is Correlated with Reduced E-cadherin in Human Breast Cancer Specimens After Chemotherapy and in Human Breast Cancer MCF-7 Cells. HORM CANC 5, 414–423 (2014). https://doi.org/10.1007/s12672-014-0199-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-014-0199-5