
Abstract
This study examined the mechanism of release of endogenous acetylcholine (ACh) in rabbit renal cortex by applying a microdialysis technique. In anesthetized rabbits, a microdialysis probe was implanted into the renal cortex and perfused with Ringer’s solution containing high potassium concentration, high sodium concentration, a Na+/K+-ATPase inhibitor (ouabain), or an epithelial Na+ channel blocker (benzamil). Dialysate samples were collected at baseline and during exposure to each agent, and ACh concentrations in the samples were measured by high-performance liquid chromatography. High potassium had no effect on renal ACh release. High sodium increased dialysate ACh concentrations significantly. Ouabain increased dialysate ACh concentration significantly. Benzamil decreased dialysate ACh concentrations significantly both at baseline and under high sodium. The finding that high potassium-induced depolarization does not increase ACh release suggests that endogenous ACh is released in renal cortex mainly by non-neuronal mechanism. Sodium ion transport may be involved in the non-neuronal ACh release.
Similar content being viewed by others


Introduction
Acetylcholine (ACh) serves as a neurotransmitter in the brain, at the autonomic ganglia, and at the parasympathetic nerve endings. Although the exact source of local ACh acting on the endothelium remains in dispute [1, 2], local ACh activates endothelial nitric oxide synthesis contributing to endothelium-dependent vasorelaxation in renal arteries [3]. In the kidney of spontaneous hypertensive rats, exogenous ACh-induced vasodilatation is reported to be impaired [4]. While the effects of exogenous ACh are not necessarily equivalent to those of endogenous ACh, the study suggests a possibility that renal endogenous ACh release is involved in the pathogenesis of hypertension. However, the mechanism of endogenous ACh release in the kidney remains largely unknown.
Maeda et al. [5] showed that cholineacetyltransferase (ChAT) mRNA is localized to the renal cortical collecting ducts and that ChAT-positive cells are principal cells. Evans et al. [6] suggested that ACh may be synthesized by non-neuronal rabbit kidney cortical cells because release of newly synthesized ACh is increased by urea in a calcium-dependent manner, but not by potassium depolarization. They observed [3H]-choline uptake and [3H]-ACh synthesis in minces of rabbit’s kidney cortex. Therefore, there is a certain non-neuronal mechanism that synthesizes ACh from choline in the rabbit’s cortical tissue. Furthermore, Williams et al. [7] reported that the action of ACh in the kidney is also mediated through alterations of the direct transport of ions. In their study, ACh increased renal plasma flow and there was a correlation between changes in renal plasma flow and sodium excretion. Therefore, renal interstitial ion concentrations may affect endogenous ACh release in the kidney.
We have developed a microdialysis technique that allows monitoring of neuronal and non-neuronal neurotransmitter releases in various organs [8–14]. We hypothesized that the mechanism of endogenous ACh release in the kidney may be elucidated by continuous monitoring of interstitial ACh concentrations in the kidney using the microdialysis technique. In the present study, we applied the microdialysis technique to the renal cortex of anesthetized rabbits and examined neuronal and non-neuronal ACh releases in the renal cortex.
Materials and methods
Animal experiments and care were conducted in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences published by the Physiological Society of Japan. The study was approved by the Animal Subject Committee of the National Cerebral and Cardiovascular Center. Thirty Japanese white rabbits (2.3–3.0 kg in weight) were used. Anesthesia was induced by intravenous injection of pentobarbital sodium (50 mg/kg) into the marginal ear vein, and maintained by intravenous infusion of a mixture of 16 mg kg−1 h−1 α-chloralose and 100 mg kg−1 h−1 urethane. Depth of anesthesia was assessed by loss of ear pinch reflex. Mechanical ventilation was conducted with 15 ml/kg of a mixture of room air and oxygen at a respiratory rate of 30 cycles/min. Systemic arterial pressure was monitored by a fluid-filled catheter inserted into the femoral artery. Esophageal temperature was maintained between 38 and 39 °C using a heating pad.
With the animal in the right lateral decubitus position, the left kidney was exposed via a retroperitoneal approach. A dialysis probe was implanted in the renal cortex as described in “Dialysis technique” below. In protocol 1 that compared ACh release in renal cortex and in myocardium, the heart was also exposed via a left lateral thoracotomy and a dialysis probe was implanted into the posterior wall of the left ventricle. In protocols 2 and 4, two dialysis probes were implanted into the renal cortex. At the end of the experiment, the animals were euthanized by an overdose of pentobarbital sodium. The left kidney was sliced and observed macroscopically to confirm that the dialysis membrane was implanted totally within the renal cortex.
Dialysis technique
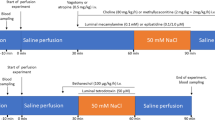
The dialysis probe used in microdialysis has been described previously [11–14]. Briefly, the probe consisted of a 13-mm dialysis fiber (PAN-1200; Asahi Chemical, Tokyo, Japan) with 25-cm polyethylene tubes attached at both ends. The dialysis probe was implanted into the renal cortex, and perfused at a speed of 2 μl/min using a microinjection pump (CMA/102, Carnegie Medicine, Sweden). The baseline perfusate was Ringer’s solution containing 100 μM eserine (Wako Pure Chemical Industries, Ltd., Osaka, Japan), a cholinesterase inhibitor. Experimental protocol was started 2 h after implantation. Four microliters of phosphate buffer (pH 3.5) was added to the sample tube before dialysate sampling. Each dialysate sampling period was 10 min, yielding a sample volume of 20 μl (Fig. 1). ACh concentration in dialysate was measured by high-performance liquid chromatography [15].

A schema of a microdialysis technique in the renal cortex. ACh acetylcholine
Experimental protocols
Protocol 1 (n = 11)
To investigate whether renal ACh is released from cholinergic nerve endings, dialysate sample was collected under high-potassium perfusion, which is widely used to examine neurotransmitter release in brain microdialysis studies [16]. In six rabbits implanted with a microdialysis probe in the renal cortex, baseline dialysate samples were collected over 10 min. After baseline sampling, the dialysis probe was perfused with a high-potassium solution (Ringer’s solution containing 200 mM KCl and 100 μM eserine) aiming to evoke depolarization of nerve terminals, and another 10-min dialysate sample was collected. For comparison, dialysate samples were collected from the posterior wall of the left ventricular myocardium under high-potassium perfusion (KCl 200 mM) in another five rabbits.
Protocol 2 (n = 6)
We investigated the effect of high sodium on renal ACh release. Two renal dialysis probes were implanted in each rabbit. After baseline dialysate sampling, each dialysis probe was perfused with Ringer’s solution containing 500 or 900 mM NaCl and 100 μM eserine. Twenty minutes after the start of high-sodium perfusion, 10-min dialysate samples were collected.
Protocol 3 (n = 7)
We investigated the effect of Na+/K+-ATPase on renal ACh release. After 10-min baseline dialysate sampling, the dialysis probe was perfused with Ringer’s solution containing a Na+/K+-ATPase inhibitor, ouabain (100 μM, Sigma-Aldrich Co. LLC., MO, USA) and 100 μM eserine. Twenty minutes after the start of ouabain perfusion, a 10-min dialysate sample was collected.
Protocol 4 (n = 6)
We investigated the effect of epithelial Na+ channel (ENaC) on renal ACh release. At baseline, one dialysis probe was perfused with Ringer’s solution containing eserine (control probe) and another probe was perfused with Ringer’s solution containing an ENaC inhibitor, benzamil (300 μM, Sigma-Aldrich Co. LLC., MO, USA) and 100 μM eserine (benzamil probe). After 10-min baseline sampling, the control probe was perfused with high-sodium Ringer’s solution (900 mM NaCl) and the benzamil probe with high-sodium Ringer’s solution (900 mM NaCl) containing 300 μM benzamil, both with the addition of 100 μM eserine. Twenty minutes after the start of high-sodium perfusion, 10-min dialysate samples were collected.
Statistical analysis
All data are presented as mean ± standard error. For each protocol, mean blood pressures at the start and end of the experiment were compared by a paired t test. In protocols 1, 2, and 3, dialysate ACh concentrations under perfusion with individual test agents were compared with baseline concentrations using a paired t test. In protocol 4, a two-way analysis of variance (ANOVA) with repeated measures on one factor (high sodium) was used to compare dialysate ACh concentrations. The other factor was benzamil. Post hoc tests were done using paired t test (the effect of high sodium) or unpaired t test (the effect of benzamil) with the significance levels corrected by Holm’s method. Statistical significance was defined as p < 0.05.
Results
Protocol 1 (n = 11)
High potassium did not affect renal dialysate ACh concentration (0.98 ± 0.19 nM at baseline vs. 0.99 ± 0.25 nM under high-potassium perfusion, not significant) (Fig. 2). Mean blood pressure did not change throughout the experiment (92.8 ± 4.1 to 92.1 ± 3.6 mmHg, not significant). On the other hand, high-potassium perfusion significantly increased cardiac dialysate ACh concentration from 2.40 ± 0.56 nM at baseline to 18.32 ± 3.53 nM (p < 0.01) (Fig. 2).

Changes of dialysate acetylcholine (ACh) concentrations in response to high-potassium perfusion (KCl 200 mM) in the renal cortex (n = 6) and in the posterior free wall of the left ventricular myocardium (n = 5). Data are expressed as mean ± standard error. **p < 0.01 vs. baseline, by paired t test
Protocol 2 (n = 6)
High sodium of 500 mM significantly increased renal dialysate ACh concentration from 1.19 ± 0.43 nM at baseline to 2.36 ± 0.44 nM (p < 0.01). High sodium of 900 mM also increased renal dialysate ACh concentration from 1.13 ± 0.29 nM at baseline to 5.01 ± 1.13 nM (p = 0.012) (Fig. 3). Mean blood pressure did not change throughout the experiment (86.2 ± 3.0 to 90.3 ± 2.8 mmHg, not significant).

Changes of dialysate acetylcholine (ACh) concentrations in response to high-sodium perfusion (NaCl 500 and 900 mM) in the renal cortex. Data are expressed as mean ± standard error (n = 6). *p < 0.05 and **p < 0.01 vs. baseline, by paired t test
Protocol 3 (n = 7)
Perfusion with ouabain significantly increased renal dialysate ACh concentration from 1.27 ± 0.22 nM at baseline to 2.25 ± 0.25 nM (p < 0.01) (Fig. 4). Mean blood pressure did not change throughout the experiment (89.7 ± 3.2 to 89.4 ± 3.6 mmHg, not significant).

Change of dialysate acetylcholine (ACh) concentration in response to perfusion of a Na+/K+-ATPase inhibitor (ouabain 100 μM) in the renal cortex. Data are expressed as mean ± standard error (n = 7). **p < 0.01 vs. baseline, by paired t test
Protocol 4 (n = 6)
In control conditions, high-sodium perfusion of 900 mM significantly increased renal dialysate ACh concentration from 0.77 ± 0.19 nM at baseline to 3.04 ± 0.46 nM. In the presence of benzamil, high-sodium perfusion also increased renal dialysate ACh concentration from 0.20 ± 0.03 nM at baseline to 0.67 ± 0.09 nM. Benzamil significantly reduced renal dialysate ACh concentration in both baseline and high-sodium conditions (high sodium, F 1,10 = 67.12, p < 0.0001; benzamil, F 1,10 = 21.83, p = 0.0009; interaction, F 1,10 = 29.00, p = 0.0003 by two-way ANOVA) (Fig. 5). Mean blood pressure did not change throughout the experiment (91.1 ± 2.0 to 93.7 ± 4.5 mmHg, not significant).

Effects of perfusion of an epithelial Na+ channel blocker (benzamil 300 μM) on dialysate acetylcholine (ACh) concentrations under baseline and high-sodium (NaCl 900 mM) conditions. Data are expressed as mean ± standard error (n = 6). *p < 0.05 and **p < 0.01, by Holm’s method
Discussion
The present study demonstrated that ACh was released by non-neuronal mechanism in the renal cortex. This non-neuronal ACh release was associated with sodium ion transport in the renal cortex. Na+/K+-ATPase and ENaC may be involved in the endogenous ACh release.
Neuronal release of endogenous ACh
When ACh is released by the neuronal mechanism, interstitial neurotransmitter concentration monitored by microdialysis increases in response to high potassium-induced depolarization of nerve terminals [8]. In protocol 1, when the microdialysis probe implanted in the left ventricular wall was perfused by a 200-mM KCl solution, dialysate ACh concentration increased significantly. This finding suggests that ACh release in the left ventricle is mainly dependent on neuronal mechanism. On the other hand, ACh concentration in the dialysate sampled from the microdialysis probe implanted in the renal cortex was not influenced by perfusion of the same KCl solution. This finding suggests that endogenous ACh release in the kidney is less dependent on neuronal mechanism compared with that in the left ventricle. Therefore, non-neuronal mechanisms may play an important role in renal endogenous ACh release.
Non-neuronal mechanism of endogenous ACh release
The present study demonstrated that an increase in interstitial sodium concentration significantly enhanced endogenous ACh release in the renal cortex. Dialysate ACh concentration during perfusion of a 500-mM NaCl solution increased two-fold compared to baseline level. Perfusion with 900-mM NaCl solution resulted in further increase in dialysate ACh concentration to fivefold the baseline level. Evans et al. [6] reported that ACh synthesis in the renal cortex was inhibited by the removal of sodium ions, suggesting that endogenous ACh release in the renal cortex may be dependent on interstitial sodium concentration. Williams et al. [7] reported that infusion of ACh into the renal artery increased fractional excretion of sodium. Takeda et al. [17] also reported that ACh caused natriuresis through inhibition of sodium reabsorption across the collecting duct cells of the renal cortical collecting ducts. Therefore, the action of ACh in the kidney may be mediated through direct alteration of ion transports.
While the results of protocol 2 indicate that extracellular high-sodium condition is involved in renal ACh release, it remains unclear whether intracellular sodium level is associated with endogenous ACh release or not. In protocol 3, addition of the Na+/K+-ATPase inhibitor ouabain in the perfusate significantly enhanced endogenous ACh release in the renal cortex. Since Na+/K+-ATPase is a ubiquitous enzyme responsible for the creation and maintenance of Na+ and K+ gradient across cell membrane by transporting 3 Na+ out and 2 K+ into the cell [18], blocking this enzyme by ouabain consequently raises intracellular sodium levels. Therefore, the present result suggests that an increase in intracellular sodium level enhances endogenous ACh release in the renal cortex. Meyer and Cooper [19] reported that inhibition of Na+/K+-ATPase elevated ACh release independent of the external calcium concentration in rat cerebral cortical synaptosomes. Blasi et al. [20] suggested that one of the mechanisms involved in ouabain-induced ACh release in the absence of Ca2+ is an increase in intracellular Na+ that evokes Ca2+ release from internal stores and inhibits ATP-dependent Ca2+ uptake in the Torpedo marmorata electric organ. This calcium-independent mechanism may partly participate in renal ACh release. In addition, we have already reported that ouabain-induced ACh release at cardiac parasympathetic nerve terminals is attributable to the mechanism of exocytosis triggered by regional depolarization [21]. However, the results of high-potassium protocol (protocol 1) indicated that depolarization of ACh-releasing cells did not contribute much to the renal ACh release observed in the present study.
As a possible mechanism for alteration of intracellular sodium level under pathophysiological conditions, we examined the role of ENaC in endogenous ACh release. In protocol 4, the ENaC blocker benzamil significantly suppressed endogenous ACh release in both baseline and high-sodium conditions. Since ENaC is a constituent of apical or outward-facing membranes of the salt-reabsorbing epithelium that transports Na+ into the cell by electrodiffusion [22], blockade of this channel causes a decrease in intracellular sodium level. Therefore, a decrease in intracellular sodium may suppress endogenous ACh release in the renal cortex. Kakizoe et al. [23] reported aberrant expression and activation of ENaC in Dahl salt-sensitive rats. Since ACh has been reported to increase sodium delivery and decrease sodium reabsorption in the collecting duct [24], impairment of endogenous ACh release despite the increase in intracellular sodium level due to augmented ENaC activity may be associated with the progression of hypertension in Dahl salt-sensitive rats. Furthermore, recent reports suggest that α7-nicotinic ACh receptors play a critical role in renal anti-inflammatory pathway. Chen et al. [25] reported that α7-nicotinic ACh receptor downregulation occurred in two-kidney one-clip hypertensive rat’s kidney. Truong et al. [26] reported that absence of α7-nicotinic ACh receptor subunit amplified inflammation and accelerated the onset of fibrosis in inflammatory kidney model mice. Therefore, endogenous ACh may exert renoprotective effects against high-sodium conditions through α7-nicotinic ACh receptor-mediated anti-inflammatory pathway.
Methodological considerations
Since ACh is degraded by ACh esterase immediately after its release, the addition of eserine into the perfusate is required for measuring in vivo release of ACh. The presence of eserine around the microdialysis fiber could have affected ACh release in the vicinity of the fiber. Notwithstanding this limitation, the microdialysis has the unique advantage of permitting elucidation of local ACh release mechanisms by delivering pharmacological agents locally without significantly affecting systemic hemodynamics. Changes in dialysate ACh concentration upon perfusion of pharmacological agents relative to baseline are considered to reflect release and disposition of local ACh.
In the present study, an increase in interstitial sodium concentration was associated with increasing the osmotic pressure of perfusate. While the results of ouabain and benzamil protocols (protocol 3 and 4) suggested that sodium ion transport was involved in renal ACh release, further investigations are required to identify the effects of osmotic pressure on renal endogenous ACh release.
Conclusions
Endogenous ACh release in the renal cortex is mainly dependent on non-neuronal mechanisms. This non-neuronal ACh release is associated with sodium ion transport in the renal cortex. In addition, Na+/K+-ATPase and ENaC may be involved in renal endogenous ACh release.
References
Kawashima K, Watanabe N, Oohata H, Fujimoto K, Suzuki T, Ishizaki Y, Morita I, Murota S (1990) Synthesis and release of acetylcholine by cultured bovine arterial endothelial cells. Neurosci Lett 119:156–158
Parnavelas JG, Kelly W, Burnstock G (1985) Ultrastructural localization of choline acetyltransferase in vascular endothelial cells in rat brain. Nature 316:724–725
Wang D, Borrego-Conde LJ, Falck JR, Sharma KK, Wilcox CS, Umans JG (2003) Contributions of nitric oxide, EDHF, and EETs to endothelium-dependent relaxation in renal afferent arterioles. Kidney Int 63:2187–2193
Tuncer M, Vanhoutte PM (1993) Response to the endothelium-dependent vasodilator acetylcholine in perfused kidneys of normotensive and spontaneously hypertensive rats. Blood Press 2:217–220
Maeda S, Jun JG, Kuwahara-Otani S, Tanaka K, Hayakawa T, Seki M (2011) Non-neuronal expression of choline acetyltransferase in the rat kidney. Life Sci 89:408–414
Evans S, Garg LC, Meyer EM (1992) Synthesis and release of acetylcholine in the rabbit kidney cortex. Life Sci 51:1699–1703
Williams RL, Pearson JE, Gonzalez FM (1982) Comparison of effects of cyanide and acetylcholine on renal hemodynamics and sodium excretion. J Pharm Sci 71:47–50
Kawada T, Yamazaki T, Akiyama T, Sato T, Shishido T, Yoshimura R, Inagaki M, Tatewaki T, Sugimachi M, Sunagawa K (2000) Local epinephrine release in the rabbit myocardial interstitium in vivo. J Auton Nerv Syst 78:94–98
Kawada T, Akiyama T, Shimizu S, Kamiya A, Uemura K, Li M, Shirai M, Sugimachi M (2009) Detection of endogenous acetylcholine release during brief ischemia in the rabbit ventricle: a possible trigger for ischemic preconditioning. Life Sci 85:597–601
Komaki F, Akiyama T, Yamazaki T, Kitagawa H, Nosaka S, Shirai M (2013) Effects of intravenous magnesium infusion on in vivo release of acetylcholine and catecholamine in rat adrenal medulla. Auton Neurosci 177:123–128
Shimizu S, Akiyama T, Kawada T, Shishido T, Yamazaki T, Kamiya A, Mizuno M, Sano S, Sugimachi M (2009) In vivo direct monitoring of vagal acetylcholine release to the sinoatrial node. Auton Neurosci 148:44–49
Shimizu S, Akiyama T, Kawada T, Shishido T, Mizuno M, Kamiya A, Yamazaki T, Sano S, Sugimachi M (2010) In vivo direct monitoring of interstitial norepinephrine levels at the sinoatrial node. Auton Neurosci 152:115–118
Shimizu S, Akiyama T, Kawada T, Sonobe T, Kamiya A, Shishido T, Tokudome T, Hosoda H, Shirai M, Kangawa K, Sugimachi M (2011) Centrally administered ghrelin activates cardiac vagal nerve in anesthetized rabbits. Auton Neurosci 162:60–65
Shimizu S, Kawada T, Akiyama T, Turner MJ, Shishido T, Kamiya A, Shirai M, Sugimachi M (2015) Guanfacine enhances cardiac acetylcholine release with little effect on norepinephrine release in anesthetized rabbits. Auton Neurosci 187:84–87
Akiyama T, Yamazaki T, Ninomiya I (1994) In vivo detection of endogenous acetylcholine release in cat ventricles. Am J Physiol 266:H854–H860
Nilsson OG, Kalén P, Rosengren E, Björklund A (1990) Acetylcholine release in the rat hippocampus as studied by microdialysis is dependent on axonal impulse flow and increases during behavioural activation. Neuroscience 36:325–338
Takeda M, Yoshitomi K, Taniguchi J, Imai M (1994) Inhibition of amiloride-sensitive apical Na+ conductance by acetylcholine in rabbit cortical collecting duct perfused in vitro. J Clin Invest 93:2649–2657
Suhail M (2010) Na+, K+-ATPase: ubiquitous multifunctional transmembrane protein and its relevance to various pathophysiological conditions. J Clin Med Res 2:1–17
Meyer EM, Cooper JR (1981) Correlations between Na+-K+ ATPase activity and acetylcholine release in rat cortical synaptosomes. J Neurochem 36:467–475
Blasi JM, Ceña V, González-García C, Marsal J, Solsona C (1988) Ouabain induces acetylcholine release from pure cholinergic synaptosomes independently of extracellular calcium concentration. Neurochem Res 13:1035–1041
Yamazaki T, Akiyama T, Kitagawa H, Komaki F, Mori H, Kawada T, Sunagawa K, Sugimachi M (2007) Characterization of ouabain-induced noradrenaline and acetylcholine release from in situ cardiac autonomic nerve endings. Acta Physiol (Oxf) 191:275–284
Garty H, Palmer LG (1997) Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77:359–396
Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, Tomita K (2009) Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens 27:1679–1689
Wilson DR, Honrath U, Sonnenberg H (1986) Effect of acetylcholine and secretin on medullary collecting duct function in the rat. Can J Physiol Pharmacol 64:62–65
Chen JK, Zhao T, Ni M, Li DJ, Tao X, Shen FM (2012) Downregulation of alpha7 nicotinic acetylcholine receptor in two-kidney one-clip hypertensive rats. BMC Cardiovasc Disord 12:38
Truong LD, Trostel J, Garcia GE (2015) Absence of nicotinic acetylcholine receptor α7 subunit amplifies inflammation and accelerates onset of fibrosis: an inflammatory kidney model. FASEB J 29:3558–3570
Acknowledgments
This study was supported by JSPS KAKENHI Grant Number 15K09110, Hiroshi and Aya Irisawa Memorial Promotion Award for Young Physiologists and grants of the Salt Science Research Foundation (No. 1427, 1539) and the Takeda Science Foundation, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
About this article

Cite this article
Shimizu, S., Akiyama, T., Kawada, T. et al. Sodium ion transport participates in non-neuronal acetylcholine release in the renal cortex of anesthetized rabbits. J Physiol Sci 67, 587–593 (2017). https://doi.org/10.1007/s12576-016-0489-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-016-0489-5