
Abstract
Ischemia induces physiological alterations in neurons that lead to cell death. This study investigated the effects of pre-ischemic exercise on CA3 neurons. Rats were divided into three groups. Animals in the exercise group were trained 5 days a week for 4 weeks. Ischemia was induced by occlusion of both common carotid arteries (CCAs) for 20 min. Apoptotic cell death was detected by TUNEL assay. Furthermore, expression of different proteins was determined by immunohistochemical staining. The number of TUNEL-positive cells was significantly increased in the ischemia group, but pre-ischemic exercise significantly reduced apoptotic cell death (P < 0.001). In addition, our results showed a significant increase in the Bax/Bcl-2 ratio in the ischemia group. Pre-ischemic exercise attenuated this ratio (P < 0.05). Furthermore, the number of active caspase-3-positive neurons was significantly increased in the ischemia group, which was reduced markedly by exercise preconditioning (P < 0.05). This study showed that pre-ischemic exercise can exert neuroprotective effects against ischemia in CA3 neurons.
Similar content being viewed by others

Introduction
Cerebral ischemia leads to a cascade of events that causes some important cellular changes [1]. Ischemia leads to selective loss of vulnerable neurons by apoptosis in specific brain regions. Certain areas of the brain as well as certain types of neurons are more sensitive to cerebral ischemia (e.g., pyramidal neurons of the hippocampus) [2, 3]. Moreover, it has been reported that apoptosis is the most important process in hippocampal neurons exposed to transient global ischemia [4]. According to the findings of previous studies, neuronal death is induced by free radical formation, brain inflammatory response and apoptosis [5]. Caspase-3 is the most widely studied member of the caspase family and is one of the key executors of apoptosis. Activation of caspase-3 is an important hallmark of apoptosis following ischemic brain insults [6].
The Bcl-2 multigene superfamily includes antiapoptotic genes such as Bcl-2, Bcl-xL and Bak [7] and proapoptotic genes such as Bax and Bad [8]. Although characterized as genes that are associated with developmental cell death, accumulating evidence suggests that these genes may participate in both pathologic apoptotic and necrotic cell death pathways [9].
Decreased immune reactivity of Bcl-2 and Bcl-xL and increased Bax immune reactivity were observed in injured neurons following both focal [10] and global ischemia [11], while the Bcl-2 and Bcl-xL levels were relatively unchanged in neurons that survived following the ischemic insult [10]. Although these observations support the hypothesis that cell survival is dependent on the ratio of anti- and proapoptotic Bcl-2 proteins [12], previous studies indicated the role of Bcl-2 and related mechanisms in the apoptosis process. Bcl-2 is a survival factor that can block both necrotic and apoptotic cell death [9]. Bcl-2 acts upstream to prevent caspase activation, inhibits free radical formation, regulates calcium sequestration [13] and blocks the proapoptotic actions of other members of the Bcl-2 family such as Bax and Bad [14].
The CA3 area holds a strategic position in the hippocampus because it receives sensory information from the external and internal environment via two main pathways: the mossy fibers and perforant path [15]. Florian et al. [16] suggested that the CA3 area is essential for spatial memory processes and specifically for memory consolidation of spatial information. A previous study demonstrated that neuronal cell death in CA3 began to appear after 6 min of ischemia [17]. Furthermore, Liu et al. showed that the expression of TUNEL-positive neurons was high in the cortical and CA3 areas of the hippocampus following cerebral ischemia/reperfusion [18].
Previous evidence indicated that physical exercise has protective effects against ischemia/reperfusion-induced injury in many organs such as the brain and heart in rats [19, 20]. Physical exercise provides this protective effect by amelioration of risk factors. Moreover, it has been shown that exercise can exert endogenous neuroprotection from ischemia/reperfusion injury by preserving neuronal viability [20, 21]. Furthermore, according to previous studies, physical exercise improves cognitive function via increased cellular oxygenation [22]. Seo et al. [23] indicated that treadmill exercise has protective effects on the hippocampal dentate gyrus granular cells in ischemic gerbils. Nevertheless, the mechanism by which physical exercise induces protection particularly in the CA3 area of the hippocampus following cerebral ischemia remains unclear.
The purpose of this study was to investigate the effects of pre-ischemic exercise on apoptosis and apoptosis-related protein (Bax, Bcl-2 and caspase-3) expression in hippocampal CA3 pyramidal neurons of male rats after induction of transient global cerebral ischemia.
Materials and methods
Animals
Twenty-one adult male Wistar rats (weighing 260–300 g) were obtained from the Tehran Pasteur Institute and housed in standard cages and a controlled environment (22–24 °C, 45–50 % humidity and 12-h light/dark cycle) with free access to food and water. All experiments were performed in accordance with the Helsinki Declaration.
Experimental groups
Rats were randomly divided into three groups: the sham operation group, exercise + ischemia group (ischemia with 4 weeks of pre-ischemic exercise) and ischemia group (7 rats in each group). In the ischemia group, the animals underwent occlusion of both common carotid arteries (CCA). In the exercise + ischemia group, the animals had 4 weeks of exercise before induction of ischemia. Sham-operated animals (serving as controls) were subjected to the same surgical procedures except that the common carotid arteries were not occluded.
Exercise training protocol
The rats in the training intervention group were trained to run on a treadmill (4-lane animal treadmill; IITC Life Science Inc., USA) 5 days a week for 4 weeks. Initially, the rats were acclimatized to run for 10–15 min at 5–7 m/min, 0 % slope for 2 days before the formal treadmill training sessions. Initially, electrical shocks (1.0 mA) were needed to force animals to run forward. Subsequently, they ran without electrical stimulation. After an adaptive running session, the rats started formal training. The rats in the training groups were scheduled to run on the treadmill for all 4 weeks. The formal treadmill training was started with a speed of 18 m/min for 35 min and 0° slope for 5 days per week in first week. The duration and intensity of the exercise and treadmill slope were increased progressively so that the animals were running for 40 min at 18 m/min with 5° slope, 45 min at 18 m/min with 10° slope and 50 min at 18 m/min with 15° slope, respectively, in the second, third and fourth week of training. Sedentary animals were placed on a stationary treadmill daily and were given electrical stimulation in a manner identical to that used for the exercise group. In order to monitor possible stress induced by treadmill running, body weight was monitored every 3 days.
Induction of transient global cerebral ischemia
To induce transient cerebral ischemia, 3 days after the last exercise training, rats were anesthetized with ketamine/xylazine (40 mg/kg, IP), then both common carotid arteries were exposed and freed from the carotid sheet, and the vagus nerves were carefully separated [24]. Both common carotid arteries were occluded for 20 min using Yasargil aneurysm micro clips. Subsequently, the carotid arteries were released and inspected for immediate reperfusion. Recirculation of blood flow was established by releasing the clips, and restoration of blood flow in the carotid arteries was confirmed by observation. Rectal temperature was maintained at 36.5 ± 0.5 °C throughout the surgery by using a feedback-regulated heating system. Animals were returned to their home cage after the surgery with free access to food and water and kept separately for 4 days (96 h).
Tissue preparation
Four days (96 h) after ischemia, the rats were anesthetized, and transcardiac perfusion was performed with 0.9 % saline, followed by 4 % paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brains were then extracted and post-fixed in the same fixative overnight. Then brains were embedded in paraffin. Afterward, 7-μm-thick paraffin-embedded coronal sections were cut (with a microtome) for TUNEL and immunohistochemical staining between 3.3 and 4.2 mm posterior to the bregma fortune according to the paxinos atlas [25].
TUNEL staining
DNA fragmentation and apoptotic cell death were detected using a TUNEL assay [26, 27]. TUNEL staining was performed using an In Situ Cell Death Detection Kit (Roche, Mannheim, Germany), a kit for the detection and quantification of apoptosis on the single cell level, based on labeling of DNA stand breaks, according to the manufacturer’s protocol. Briefly, the sections (three sections per animal) were deparaffinized in xylene (Sigma), rehydrated by successive series of alcohol, washed in PBS and permeabilized by proteinase K 10 mM for 30 min at room temperature. Then, the sections were rinsed and incubated with 3 % H2O2 in methanol for 10 min in the dark to block the endogenous peroxidase. Then the sections were incubated in the TUNEL reaction mixture for 60 min at 37 °C in humidified atmosphere. After washing the sections with PBS, sections were visualized by using a converter-POD for 30 min at 37 °C in a humidified atmosphere in the dark, then rinsed with PBS and 50–100 μl DAB (0.05 % 3,3-diaminobenzidine) substrate as a chromogen was added for 10 min and rinsed with PBS. Then all slides were mounted on a coverslip and were analyzed by light microscope (Olympus AX-70). TUNEL-positive cells were quantified using light microscopy at 400× magnification. Images were analyzed using Image Tool 2 software. The number of TUNEL-positive cells was counted along a 400-µm-long transect (0.160 mm2) of the CA3 area of the right hippocampus. All counting procedures were performed blindly.
Immunohistochemical staining
The purpose of this study was to investigate levels of different proteins (Bax, Bcl-2 and caspase-3) in the CA3 area of the hippocampus. We are faced with technical limitations in separating the CA3 area from other parts of the hippocampus for RT-PCR and Western blot analysis; we used immunohistochemical staining to better detect protein expression. Immunohistochemical staining was performed on 7-μm tissue sections (three sections per animal) [28]. Briefly, tissue sections were incubated for 30 min at 60 °C, rehydrated through a descending alcohol series and treated with 10 % hydrogen peroxide in methanol for 10 min to reduce endogenous peroxidase activity. After being washed in Tris buffer [H2NC (CH2OH)3, pH 7.4], antigens were retrieved by autoclaving for 11 min in citrate buffer (C6H5Na3O7·2H2O, pH 6). Then, sections were incubated with primary antibodies (Biorbyt, UK) overnight at 4 °C temperature. Optimal dilution was found to be 1/100. Tissues were then incubated in goat polyclonal secondary antibody (HRP) (Biorbyt, UK) for 30 min at room temperature with the addition of DAB (Sigma, USA) to achieve visualization of the antigen. In the final step, tissue sections were lightly counterstained with hematoxylin (Sigma), dehydrated in alcohol, cleared in xylene and mounted for visualization. The primary antibody was not included in the processing of the negative control slide. Human cervical carcinoma tissue was used as a positive control. Photomicrographs of sections were prepared using light microscopy at 400× magnification by a blinded investigator. The number of immune positive cells was counted along the 400-µm-long transect (0.160 mm2) of the CA3 area of the right hippocampus.
Statistical analysis
All results are reported as mean ± SD. The Kolmogorov-Smirnov test was used to verify the normality of the distribution. One-way analysis of variance (ANOVA) test was used to compare the differences between the groups. When a significant difference was revealed, the Dunnett’s T3 or Scheffé’s post hoc test was used to specify where the difference occurred. When the homogeneity of variance was established, Scheffé’s post hoc test was used; otherwise, we applied Dunnett’s T3 post hoc test. The level of significance was set at P < 0.05. All data were analyzed using the SPSS software package (SPSS for Windows; SPSS Inc., Chicago, IL, USA; version 16.00).
Results
Pre-ischemic exercise reduced ischemia/reperfusion-induced apoptosis in the CA3 area following ischemia
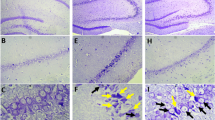
The results of TUNEL staining showed that the number of TUNEL-positive cells in the CA3 area of the hippocampus in the ischemia group significantly increased (12.2 ± 2.4) compared to the sham group (1.8 ± 0.5, P < 0.001), indicating that transient cerebral ischemia induces apoptosis in hippocampal CA3 pyramidal cells. On the other hand, in ischemic rats preconditioned with exercise, the number of TUNEL-positive cells was decreased (7.0 ± 1.2) compared to the ischemia group (P < 0.001) (Figs. 1, 2).

Photomicrographs of TUNEL staining in the hippocampus after transient global cerebral ischemia. a The CA3 area of the hippocampus, b sham-operated group, c ischemia group and d exercise + ischemia group (arrows indicate the active TUNEL-positive cells, magnification ×400)

Effects of exercise preconditioning on the number of hippocampal CA3 area TUNEL-positive cells after ischemia. *Significantly different compared with the sham group (P < 0.001). #Significantly different compared with the sham (P < 0.01) and ischemia (P < 0.001) groups
Pre-ischemic exercise increased the Bcl-2 protein level in the CA3 area
According to the results of Bcl-2 immunohistochemical staining, there was a significant difference in the number of Bcl-2-positive cells among groups in the CA3 area. Bcl-2 protein expression was higher in the sham-operated group (27.0 ± 1.4), and ischemia decreased the number of Bcl-2-positive cells (16.2 ± 2.8, P < 0.01). In ischemic rats preconditioned with exercise, the number of Bcl-2-positive cells increased (27.1 ± 3.7) compared to the ischemia group (P < 0.001) (Figs. 3, 4).

Photomicrographs of Bcl-2 immunohistochemical staining in the CA3 area of right hippocampus after transient global cerebral ischemia. a The CA3 area of the hippocampus, b sham-operated group, c ischemia group and d exercise + ischemia group (arrows indicate the Bcl-2-positive cells, magnification ×400)

Effects of exercise preconditioning on the number of Bcl-2-positive cells in the rats’ hippocampal CA3 area after the transient global cerebral ischemia. Exercise preconditioning significantly attenuated the ischemia/reperfusion-induced increase in the number of Bcl-2-positive cells. *Significantly different compared with the sham group (P < 0.01). #Significantly different compared with the ischemia group (P < 0.001)
Effect of exercise preconditioning on the Bax protein level in the CA3 area following ischemia
The results of Bax immunohistochemical staining showed that there was no notable difference among groups in the number of Bax-positive cells in the CA3 area. Bax protein expression in the ischemia group (27.4 ± 10.4) was not significantly different compared to the sham-operated group (11.5 ± 3.5, P > 0.05). In addition, physical exercise (23.5 ± 5.2) did not change the level of this protein significantly compared to the ischemia group (P > 0.05) (Figs. 5, 6).

Photomicrographs of Bax immunohistochemical staining in the CA3 area of the right hippocampus after transient global cerebral ischemia. a The CA3 area of the hippocampus, b sham-operated group, c ischemia group and d exercise + ischemia group (arrows indicate the Bax-positive cells, magnification ×400)

Effects of exercise preconditioning on the number of Bax-positive cells in the rats’ hippocampal CA3 area after transient global cerebral ischemia. Exercise did not cause significant changes in the number of Bax-positive cells
Exercise preconditioning modulated the Bax/Bcl-2 ratio in the CA3 area following ischemia
The Bax/Bcl-2 ratio was significantly different among groups. The Bax/Bcl-2 ratio was higher in the ischemia group (1.6 ± 0.6) compared to the sham-operated group (0.4 ± 0.1, P < 0.05) and was lower in rats preconditioned with exercise (0.8 ± 0.2) (P < 0.05) (Fig. 7).

Effects of exercise preconditioning on the Bax/Bcl-2 ratio in the rats’ hippocampal CA3 neurons after transient global cerebral ischemia. Exercise preconditioning significantly modulated the ischemia/reperfusion-induced increase in the Bax/Bcl-2 ratio. *Significantly different compared with the sham group (P < 0.05). #Significantly different compared with the ischemia group (P < 0.05)
Physical exercise reduced ischemia-induced caspase-3 activation
According to the results of caspase-3 immunohistochemical staining, there was a significant difference in the number of active caspase-3-positive cells among groups. The number of active caspase-3-positive cells was significantly higher in the ischemia group (17.0 ± 3.4) compared to the sham-operated group (4.0 ± 0.8, P < 0.001), while the rats preconditioned with exercise had fewer active caspase-3-positive cells in the CA3 area (P < 0.05) (Figs. 8, 9).

Photomicrographs of caspase-3 immunohistochemical staining in the CA3 area of the right hippocampus after transient global cerebral ischemia. a The CA3 area of the hippocampus, b sham-operated group, c ischemia group and d exercise + ischemia group (arrows indicate the active caspase-3 positive cells, magnification ×400)

Effects of exercise preconditioning on the number of active caspase-3-positive cells in the rat’s hippocampal CA3 area after transient global cerebral ischemia. Exercise preconditioning significantly attenuated the ischemia/reperfusion-induced caspase-3 activation. *Significantly different compared with the sham group (P < 0.001). #Significantly different compared with the sham (P < 0.01) and ischemia (P < 0.05) groups
Discussion
In this study, we investigated the effects of pre-ischemic exercise on apoptosis and apoptosis-related protein (Bax, Bcl-2 and Caspase-3) expression in the CA3 area of the hippocampus following transient global cerebral ischemia. The results of TUNEL staining demonstrated that the number of TUNEL-positive cells in the CA3 area of the hippocampus was significantly higher following cerebral ischemia. However, treadmill exercise significantly suppressed the ischemia-induced increase in apoptotic cell death in this area; in addition, ischemic insult enhanced the Bax/Bcl-2 ratio in the hippocampal CA3 area. Also, pre-ischemic exercise, by increasing Bcl-2, with no change in the Bax levels, significantly attenuated the Bax/Bcl-2 ratio. Moreover, the results of caspase-3 immunohistochemical staining demonstrated that the number of active caspase-3-positive cells in the CA3 area of the hippocampus was significantly increased following transient global cerebral ischemia. Pretreatment with exercise significantly reduced the ischemia/reperfusion-induced caspase-3 activation.
Our findings support the hypothesis that modulation of alterations in the Bax/Bcl-2 ratio with treadmill exercise can regulate downstream caspase-driven apoptosis following transient global cerebral ischemia. The underlying mechanism by which this action takes place is still unknown. It has been well established that caspase signaling, inflammatory factors, oxidative stress, calcium dysregulation and excitotoxicity represent the main causes of neuronal apoptosis [29]. It seems any factor that inhibits this process can be used in the treatment of brain ischemia.
One of the possible mechanisms of the neuroprotective effect of exercise in the CA3 area following transient global ischemia/reperfusion is the capacity to block NMDA-induced cytotoxicity. NMDA receptor-mediated intracellular calcium ([Ca2+]i) elevation, as well as subsequently high levels of Ca2+ within the mitochondria, has an essential role in mitochondrial membrane depolarization, modulating its permeability. This is an important step in the process of caspase activation and apoptosis [30]. Previous studies have shown that cerebral ischemia can lead to the uncontrolled release of glutamate [31]. Overactivation or misuse of glutamate and its receptors has been shown to increase injury following cerebral ischemia, especially in neurons of the hippocampus [32]. According to previous studies, exercise preconditioning may lessen the overexpression of glutamate and be involved in glutamate receptor downregulation, consequently reducing brain damage after ischemia [33]. Jia et al. [31] showed in their study that treadmill training can protect striatal neurons from ischemic injury. They suggested that this protective effect takes place by preventing NMDA-induced cytotoxicity [31].
It is known that reperfusion injury plays an important role in the brain ischemic cascade [34]. Returned blood flow can reintroduce oxygen to the cells, damaging their proteins, DNA and plasma membranes. The level of free radicals may increase if the plasma membrane is damaged. These processes can play an important role in redox signaling, ultimately causing cell apoptosis [35]. Reactive oxygen species (ROS) are produced in the mitochondrial electron transport chain as a normal product, but when their level exceeds the cellular antioxidant capacity, they can lead to cell death. ROS-induced oxidative stress plays a crucial role in the ischemic cascade [36, 37]. According to previous studies, Bcl2 protein is known to be a factor that can enhance the antioxidant activity by increasing the GSH level and Cu/Zn superoxide dismutase (SOD1) activity [38]. Therefore, another possible mechanism for the neuroprotective effect of exercise in the CA3 area of the hippocampus following cerebral ischemia could be related to the increased level of Bcl-2 and its ability to block free radical formation.
Extracellular signal-regulated kinase 1/2 (ERK1/2) are involved in mitogen-activated protein kinase pathways and are constitutively expressed in the adult brain [39]. These ERK1/2-regulated pathways are very important in signal transduction and neuroprotection in the setting of ischemia/reperfusion injury. Based on the results of previous studies, activation of this regulatory kinase enables tissue repair in the setting of ischemia/reperfusion injury, which results in a decrease in cell death [40].
Furthermore, exercise preconditioning appears to upregulate ERK1/2 [41]. In addition, a recent study demonstrated that pre-ischemic treadmill training, by upregulation of heat shock protein-70 (HSP-70), can reduce the brain infarction volume. Moreover, HSP-70 and ERK1/2 can increase Bcl-2 protein and decrease the Bax/Bcl-2 ratio [37].
Therefore, Bcl-2, by blocking free radical formation, regulating calcium sequestration, preventing caspase-3 activation and blocking the proapoptotic actions of other members of the Bcl-2 family such as Bax and Bad, ultimately can inhibit apoptosis following transient global cerebral ischemia in the hippocampal CA3 area neurons.
In conclusion, our study demonstrated that pre-ischemic treadmill training results in the reduction of the Bax/Bcl-2 ratio and caspase-3 activity in the hippocampal CA3 neurons against cerebral ischemia. Physical exercise can exert protective effects in brain ischemia models via some possible mechanisms such as preventing NMDA receptor cytotoxicity, reducing ROS production and upregulating ERK1/2 and HSP-70. The neuroprotective mechanisms of exercise can provide a neuroprotective therapy that will simultaneously promote cell survival and decrease neuronal death, therefore ameliorating much of the functional and memory loss following ischemic stroke.
References
Lee J-M, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399:A7–A14
Mitani A, Andou Y, Kataoka K (1992) Selective vulnerability of hippocampal CA1 neurons cannot be explained in terms of an increase in glutamate concentration during ischemia in the gerbil: brain microdialysis study. Neuroscience 48:307–313
Erfani S, Khaksari M, Oryan S, Shamsaei N, Aboutaleb N, Nikbakht F et al (2015) Visfatin reduces hippocampal CA1 cells death and improves learning and memory deficits after transient global ischemia/reperfusion. Neuropeptides 49:63–68
Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z et al (1997) Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci 94:2007–2012
Matsuo Y, Kihara T, Ikeda M, Ninomiya M, Onodera H, Kogure K (1995) Role of neutrophils in radical production during ischemia and reperfusion of the rat brain: effect of neutrophil depletion on extracellular ascorbyl radical formation. J Cereb Blood Flow Metab 15:941–947
Hwang L, Choi I-Y, Kim S-E, Ko I-G, Shin M-S, Kim C-J et al (2013) Dexmedetomidine ameliorates intracerebral hemorrhage-induced memory impairment by inhibiting apoptosis and enhancing brain-derived neurotrophic factor expression in the rat hippocampus. Int J Mol Med 31:1047–1056
Chittenden T, Harrington EA, O’Connor R, Remington C, Lutz RJ, Evan GI et al (1995) Induction of apoptosis by the Bcl-2 homologue Bak 374:733–736
Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell 80:285–291
Bredesen DE (1995) Neural apoptosis. Ann Neurol 38:839–851
Gillardon F, Lenz C, Waschke K, Krajewski S, Reed J, Zimmermann M et al (1996) Altered expression of Bcl-2, Bcl-X, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Mol Brain Res 40:254–260
Chen J, Zhu RL, Nakayama M, Kawaguchi K, Jin K, Stetler RA et al (1996) Expression of the apoptosis-effector gene, Bax, is up-regulated in vulnerable hippocampal CA1 neurons following global ischemia. J Neurochem 67:64–71
Korsmeyer SJ (1995) Regulators of cell death. Trends Genet 11:101–105
MacManus JP, Linnik MD (1997) Gene expression induced by cerebral ischemia: an apoptotic perspective. J Cereb Blood Flow Metab 17:815–832
Merry D, Korsmeyer S (1997) Bcl-2 gene family in the nervous system. Annu Rev Neurosci 20:245–267
Dolorfo CL, Amaral DG (1998) Entorhinal cortex of the rat: topographic organization of the cells of origin of the perforant path projection to the dentate gyrus. J Comp Neurol 398:25–48
Florian C, Roullet P (2004) Hippocampal CA3-region is crucial for acquisition and memory consolidation in Morris water maze task in mice. Behav Brain Res 154:365–374
Smith M-L, Auer R, Siesjö B (1984) The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol 64:319–332
Liu G, Wang T, Wang T, Song J, Zhou Z (2013) Effects of apoptosis-related proteins caspase-3, Bax and Bcl-2 on cerebral ischemia rats. Biomed Rep 1:861–867
Hao Z, Pan S-S, Shen Y-J, Ge J (2014) Exercise preconditioning-induced late phase of cardioprotection against exhaustive exercise: possible role of protein kinase C delta. J Physiol Sci 64:333–345
Liebelt B, Papapetrou P, Ali A, Guo M, Ji X, Peng C et al (2010) Exercise preconditioning reduces neuronal apoptosis in stroke by up-regulating heat shock protein-70 (heat shock protein-72) and extracellular-signal-regulated-kinase 1/2. Neuroscience 166:1091–1100
Moghaddasi M, Javanmard SH, Reisi P, Tajadini M, Taati M (2014) The effect of regular exercise on antioxidant enzyme activities and lipid peroxidation levels in both hippocampi after occluding one carotid in rat. J Physiol Sci 64:325–332
Endo K, Matsukawa K, Liang N, Nakatsuka C, Tsuchimochi H, Okamura H et al (2013) Dynamic exercise improves cognitive function in association with increased prefrontal oxygenation. J Physiol Sci 63:287–298
Seo T-B, Kim T-W, Shin M-S, Ji E-S, Cho H-S, Lee J-M et al (2014) Aerobic exercise alleviates ischemia-induced memory impairment by enhancing cell proliferation and suppressing neuronal apoptosis in hippocampus. Int Neurourol J 18:187–197
Erfani S, Aboutaleb N, Oryan S, Shamsaei N, Khaksari M, Kalalian-Moghaddam H et al (2015) Visfatin inhibits apoptosis and necrosis of hippocampus CA3 cells following transient global ischemia/reperfusion in rats. Int J Pept Res Ther 21:223–228
Paxinos G, Watson C. The rat brain in stereotaxic coordinates: hard cover edition: Academic press, London; 2006
Aboutaleb N, Kalalianmoghaddam H, Eftekhari S, Shahbazi A, Abbaspour H, Khaksari M (2014) Apelin-13 inhibits apoptosis of cortical neurons following brain ischemic reperfusion injury in a transient model of focal cerebral ischemia. Int J Pept Res Ther 20:127–132
Gheibi S, Aboutaleb N, Khaksari M, Kalalian-Moghaddam H, Vakili A, Asadi Y et al (2014) Hydrogen sulfide protects the brain against ischemic reperfusion injury in a transient model of focal cerebral ischemia. J Mol Neurosci 54:264–270
Erfani S, Khaksari M, Oryan S, Shamsaei N, Aboutaleb N, Nikbakht F (2015) Nampt/PBEF/visfatin exerts neuroprotective effects against ischemia/reperfusion injury via modulation of Bax/Bcl-2 ratio and prevention of caspase-3 activation. J Mol Neurosci 56(1):237–243
Moskowitz MA, Lo EH, Iadecola C (2010) The science of stroke: mechanisms in search of treatments. Neuron 67:181–198
Khaksari M, Aboutaleb N, Nasirinezhad F, Vakili A, Madjd Z (2012) Apelin-13 protects the brain against ischemic reperfusion injury and cerebral edema in a transient model of focal cerebral ischemia. J Mol Neurosci 48:201–208
Jia J, Hu Y-S, Wu Y, Yu H-X, Liu G, Zhu D-N et al (2010) Treadmill pre-training suppresses the release of glutamate resulting from cerebral ischemia in rats. Exp Brain Res 204:173–179
Zhang F, Wu Y, Jia J (2011) Exercise preconditioning and brain ischemic tolerance. Neuroscience 177:170–176
Zhang F, Wu Y, Jia J, Hu Y-S (2010) Pre-ischemic treadmill training induces tolerance to brain ischemia: involvement of glutamate and ERK1/2. Molecules 15:5246–5257
White BC, Sullivan JM, DeGracia DJ, O’Neil BJ, Neumar RW, Grossman LI et al (2000) Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci 179:1–33
Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA et al (1997) Early detection of DNA strand breaks in the brain after transient focal ischemia: implications for the role of DNA damage in apoptosis and neuronal cell death. J Neurochem 69:232–245
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21:2–14
Ohtsuka K, Suzuki T (2000) Roles of molecular chaperones in the nervous system. Brain Res Bull 53:141–146
Lee M, Hyun D-H, Marshall K-A, Ellerby LM, Bredesen DE, Jenner P et al (2001) Effect of overexpression of BCL-2 on cellular oxidative damage, nitric oxide production, antioxidant defenses, and the proteasome. Free Radic Biol Med 31:1550–1559
Sharony R, Pintucci G, Saunders PC, Grossi EA, Baumann FG, Galloway AC et al (2006) Matrix metalloproteinase expression in vein grafts: role of inflammatory mediators and extracellular signal-regulated kinases-1 and-2. Am J Physiol Heart Circ Physiol 290:H1651–H1659
Cavanaugh JE (2004) Role of extracellular signal regulated kinase 5 in neuronal survival. Eur J Biochem 271:2056–2059
Jones NM, Bergeron M (2004) Hypoxia-induced ischemic tolerance in neonatal rat brain involves enhanced ERK1/2 signaling. J Neurochem 89:157–167
Acknowledgments
This research was supported by a grant (contract no. 91052159) sponsored by the Iran National Science Foundation (INSF). The authors are very grateful to the INSF for financial support.
Conflict of interest
Nahid Aboutaleb, Nabi Shamsaei, Mehdi Khaksari, Sohaila Erfani, Hamid Rajabi and Farnaz Nikbakht declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Aboutaleb, N., Shamsaei, N., Khaksari, M. et al. Pre-ischemic exercise reduces apoptosis in hippocampal CA3 cells after cerebral ischemia by modulation of the Bax/Bcl-2 proteins ratio and prevention of caspase-3 activation. J Physiol Sci 65, 435–443 (2015). https://doi.org/10.1007/s12576-015-0382-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12576-015-0382-7