
Abstract
Heart failure is a life-threatening condition that carries a considerable emotional and socio-economic burden. As a result of the global increase in the ageing population, sedentary life-style, increased prevalence of risk factors, and improved survival from cardiovascular events, the incidence of heart failure will continue to rise. Despite the advances in current cardiovascular therapies, many patients are not suitable for or may not benefit from conventional treatments. Thus, more effective therapies are required. Transforming growth factor (TGF) β family of cytokines is involved in heart development and dys-regulated TGFβ signalling is commonly associated with fibrosis, aberrant angiogenesis and accelerated progression into heart failure. Therefore, a potential therapeutic pathway is to modulate TGFβ signalling; however, broad blockage of TGFβ signalling may cause unwanted side effects due to its pivotal role in tissue homeostasis. We found that leucine-rich α-2 glycoprotein 1 (LRG1) promotes blood vessel formation via regulating the context-dependent endothelial TGFβ signalling. This review will focus on the interaction between LRG1 and TGFβ signalling, their involvement in the pathogenesis of heart failure, and the potential for LRG1 to function as a novel therapeutic target.
Similar content being viewed by others
Introduction
Heart failure is a progressive and chronic condition in which the heart is no longer able to circulate blood efficiently to meet the body’s demands (Johnson 2014). A wide range of conditions such as ischemic heart disease (IHD), hypertension, valvular heart disease, myocarditis, diabetes and cardiomyopathy can lead to heart failure (Nishimura et al. 2014). In response to stress or injury, the myocardium undergoes a series of pathological changes including structural rearrangement and morphological changes of cardiomyocytes, inflammation, extracellular matrix (ECM) remodelling, microvascular rarefaction and chamber dilation (Manabe et al. 2002; Kehat and Molkentin 2010). These changes cause further deterioration in cardiac function and eventually lead to heart failure (Cohn et al. 2000).
Over the last few decades, the prevalence and incidence of heart failure continues to rise mainly due to the prolonged longevity, improved survival rate from other cardiovascular events (e.g., myocardial infarction, valvular disease, and arrhythmias), sedentary life style, and the increased prevalence of risk factors (e.g., hypertension, diabetes and obesity) (Mann DL 2012). In 2010, more than 41 million people lived with heart failure worldwide (Forouzanfar et al. 2013). Despite improved understanding of the molecular mechanisms and significant advances in treatment strategies, heart failure still carries substantial morbidity and mortality and its therapy remains a major unmet medical need. In this review, we summarise the current knowledge on the role of TGFβ1 and its novel modulator, LRG1, in different pathologies of cardiac remodelling and the potential of LRG1-targeted therapeutics for the treatment of heart failure.
The challenges of current treatments for heart failure
Heart failure is a heterogeneous disease with a broad spectrum of symptoms. Current treatments aim to alleviate symptoms, slow disease progression and thereby improve overall quality of life and survival. For example, IHD-induced heart failure, the most common type of the disease, is normally treated with antiplatelet drugs, anticoagulants and β-blockers. Emergent reperfusion via surgical or catheter-based revascularisation procedures is used to restore blood flow and improve survival following ischemic episodes (Heuser et al. 2000; Horvath 2000). However, a substantial portion of patients are not suitable for or do not benefit from conventional revascularisation treatments because of a poor overall health status or the presence of comorbidities (Norgren et al. 2007). Even in patients who received successful primary revascularisation, stent thrombosis and saphenous vein bypass graft disease can occur and cause recurrent myocardial ischemia and cardiac remodelling (Kaul et al. 1991). Furthermore, none of these treatments alters the natural history of heart failure and therefore offers no cure.
Both human and animal studies have shown that individuals with robust collateral circulation and microvascular perfusion are associated with delayed myocardial cell death (Antoniucci et al. 2002), reduced occurrence of myocardial infarction (MI) (Choi et al. 2013), smaller infarction size (Habib et al. 1991) and increased survival (Meier et al. 2012). However, the capacity of collateral and capillary vessel remodelling under ischemic condition is highly variable among individuals. Accelerating this innate physiological response by exogenous angiogenic factors has been considered as an attractive approach to bypass occluded vessels, revascularise ischemic tissues and restore tissue function (Carmeliet and Jain 2011). An impressive body of pre-clinical evidence has demonstrated improved myocardial perfusion and function upon therapeutic angiogenesis in animal models (Harada et al. 1994; Unger et al. 1994; Landau et al. 1995; Lazarous et al. 1996; Shou et al. 1997; Lopez et al. 1998; Lee et al. 2000; Zhang et al. 2002; Cao et al. 2005; Heinl-Green et al. 2005; Cao 2009). Initial phase I clinical trials in patients with advanced IHD, but who did not meet the criteria for standard revascularisation strategies, have also demonstrated an improved cardiac circulation and function after being treated with pro-angiogenic factors such as vascular endothelial growth factors (VEGF) and basic fibroblast growth factor (bFGF) (Losordo et al. 1998, 2002; Schumacher et al. 1998; Rosengart et al. 1999; Symes et al. 1999; Hendel et al. 2000; Udelson et al. 2000; Henry et al. 2001; Vale et al. 2001; Fortuin et al. 2003; Reilly et al. 2005). However, caution is needed in the interpretation of outcomes of these studies as most of them lack proper placebo controls. Not surprisingly, similar therapeutic efficacy has not yet been achieved in larger, placebo-controlled, late-stage clinical trials, which is partly due to the extent of angiogenesis observed in the placebo group (Grines et al. 2002, 2003; Simons et al. 2002; Kastrup et al. 2011).
Angiogenesis is a tightly controlled process involving multiple levels of interactions between a wide variety of molecules, cells and extracellular matrix (ECM) proteins. It is now widely accepted that a single angiogenic factor may not be sufficient to induce the formation of functional vasculatures. Indeed, the treatment of VEGF leads to the formation of leaky, chaotic and tortuous vessels that lack the normal hierarchical structure (Nagy et al. 2007; Hedlund et al. 2009; Cao et al. 2010). Furthermore, both VEGF (Thurston 2002) and bFGF (Cuevas et al. 1991) are involved in vessel dilation and their treatment is associated with severe hypotension (Hariawala et al. 1996; Horowitz et al. 1997; Unger et al. 2000; Henry et al. 2001). In addition, there is evidence that VEGF exerts detrimental pro-atherogenic effects by influencing endothelial and immune cell function (Ross 1993; Inoue et al. 1998; Kim et al. 2001). A combination treatment targeting growth factors with complementary mechanisms might be more effective and has less unwanted side effects.
ECM is essential for proper cardiac function. It provides a scaffold for different types of cells in myocardium and transmits mechanical force and signals to myocardial fibres (Banerjee et al. 2006). ECM remodelling is a critical step that allows the ordered replacement of damaged cells after injury. However, chronic inflammation and repetitive injury can cause disturbed ECM homeostasis and fibrosis, a feature shared by many conditions associated with heart failure (Weber et al. 1995). Cardiac fibrosis exaggerates mechanical stiffness of the myocardium and its vasculature, impairs myocyte contractility, disrupts electrical coupling, destroys normal tissue architecture and eventually leads to heart failure (Lopez et al. 2001; Ho et al. 2010; Karagueuzian 2011). Increasing evidence shows that fibrosis is a dynamic and reversible process (Iredale 2007). Targeting fibrosis, therefore, presents a promising strategy to prevent or slow down the deterioration of cardiac function. Despite its huge impact on cardiovascular diseases and intensive research efforts to explore new therapies, there is no approved treatment that directly targets the mechanisms of fibrosis in the heart.
TGFβ1 and heart failure
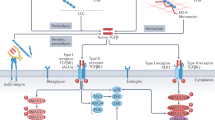
The TGFβ family of cytokines plays important roles in embryogenesis, tissue homeostasis and regeneration (Massague 2012). Their secretion, activation and function are tightly controlled by multiple mechanisms to ensure precise signal propagation (Fig. 1). TGFβs are secreted in latent form as part of a large protein complex (Khalil 1999) and their activation requires functional and physical cooperation of mannose-6-phosphate (M6P)/insulin-like growth factor II receptor (IGFIIR), urokinase-type plasminogen activator receptor (UPAR), Neuropilin 1 (NRP1) and different proteases and metalloproteases (MMPs) (Dennis and Rifkin 1991; Scott and Firth 2004; Glinka et al. 2011; Shi et al. 2011). Once released, TGFβs bind to type II receptor TGFβRII, which recruits type I receptor, activin receptor-like kinase (ALK) (Shi and Massague 2003), and activates a multitude of intracellular signalling including canonical Smad and non-canonical ERK, JNK, TAK1, P38 and Rho cascades (Derynck and Zhang 2003). TGFβs also interact extensively with other signalling pathways leading to very different even opposite outcomes (Massague 2012).

Schematic representation of TGFβ signalling and crosstalk with other signalling pathways. TGFβ ligands are synthesised as a large latent complex consisting of TGFβ dimmer covalently associated with a latency-associated peptide (LAP) and a latent TGFβ-binding protein (LTBP). The activation of latent TGFβ requires functional and physical cooperation of M6P/IGFIIR, UPAR, NRP1 and other proteases and MMPs. The released TGFβ dimers bind the type II TGFβ receptor (RII) first, which recruits and transphosphorylates the type I receptors (RI). RI propagates the signal into the cell by phosphorylating TGFβ receptor-regulated SMADs (R-Smads). They form heteromeric complexes with the common SMAD (co-Smad) and translocate to the nucleus. The R-Smads–co-Smad complex formation can be inhibited by inhibitory Smad (I-Smad). Once in the nucleus, the R-SMAD–co-SMAD complex associates with other DNA-binding transcription factors to modulate the expression of target genes. In the non-canonical pathways, the activated transforming growth factor-β (TGFβ) receptor complex transmits a signal through other factors, such as TGFβ-activated kinase 1 (TAK1), p38 mitogen-activated protein kinase (p38 MAPK), RHO, phosphoinositide 3-kinase (PI3K)–AKT, extracellular signal-regulated kinase (ERK), Rho-associated protein kinase (ROCK), or JUN N-terminal kinase (JNK). TGFβ signalling interacts extensively with other pathways, such as the WNT, Notch, AngII, IGF and VEGF pathways, which defines the context-dependent TGFβ signalling
In mammals, there are three different isoforms: TGFβ1, TGFβ2, and TGFβ3. Each of them shows distinct expression pattern and functions. TGFβ1, the focus of this review, is the predominant and most ubiquitously expressed isoform (Millan et al. 1991). In the heart, TGFβ1 regulates the signalling and function of different types of cells, including endothelial cells (ECs), vascular mural cells (pericytes in capillaries and vascular smooth muscle cells (VSMCs) in larger vessels), myofibroblasts, macrophages and cardiomyocytes (Bujak and Frangogiannis 2007; Koitabashi et al. 2011). Aberrant TGFβ1 signalling contributes to the development of a multitude of conditions associated with heart failure such as dilated and hypertrophic cardiomyopathies, post-infarction myocardial remodelling, valvular diseases and arrhythmia in both mice and humans (Cambien et al. 1996; Schultz Jel et al. 2002; Euler-Taimor and Heger 2006; Khan and Sheppard 2006; Kapur et al. 2013).
TGFβ1 and cardiac fibrosis
TGFβ1 is a potent fibrogenic factor that mediates ECM homeostasis through different mechanisms, for example, by inducing ECM (such as collagens and fibronectin) synthesis via both canonical and non-canonical signalling cascades (Chen et al. 2000; Qiao et al. 2005; Leask 2007), decreasing the production of proteinase regulating ECM degradation (such as MMPs), promoting the production of inhibitors of these proteases (such as TIMPs) (Biernacka et al. 2011) and promoting integrin expression to increase the adhesion of cells to matrix (Thannickal et al. 2003). Studies have shown that TGFβ1 signalling pathway components, including TGFβ1, ENG and Smads, are markedly up-regulated at the site of injury after MI (Hao et al. 1999; Krum et al. 2002; Dean et al. 2005; Kapur et al. 2012), in patients suffering from hypertrophic cardiomyopathy (Villarreal and Dillmann 1992, Li et al. 1998) and dilated cardiomyopathy (Pauschinger et al. 1999; Sanderson et al. 2001), and all these conditions are characterised by excessive fibrosis in the heart. Consistently, TGFβ1 overexpression in transgenic mice leads to myocardial fibrosis (Rosenkranz et al. 2002; Seeland et al. 2002). Studies have shown that TGFβ1-mediated endothelial-to-mesenchymal transition (EndoMT) also contributes to myocardial fibrosis (Zeisberg et al. 2007; van Meeteren and ten Dijke 2012). Recently, ENG, a previously considered EC specific TGFβ1 receptor, has been found to be expressed in cardiac fibroblasts and to mediate the pro-fibrotic effect of angiotensin II (AngII) via angiotensin II receptor type 1 (AT1) (Chen et al. 2004). Reduced ENG activity led to attenuated cardiac fibrosis and increased survival in an in vivo model of heart failure (Kapur et al. 2012). This study demonstrated that the expression of ENG is significantly up-regulated in human failing left ventricles. Inhibition of the activity of ENG-attenuated TGFβ1 induced Smad2/3 phosphorylation, ECM deposition and cardiac fibrosis. Eng +/− mice with pressure overload-induced heart failure showed an increased capillary density in the heart, preserved cardiac function and improved survival. Thus, targeting the TGFβ1 signalling pathway might provide an attractive strategy to limit structural deterioration of myocardium and ultimately lead to improved cardiac function, and survival of heart failure patients.
TGFβ1 and cardiomyocyte hypertrophy
In addition to fibrosis, hypertrophic growth of cardiomyocytes occurs in response to haemodynamic overload and represents the heart’s effort to maintain cardiac output sufficient to meet the body’s demands (Glennon et al. 1995). There is compelling evidence that TGFβ1 plays a critical role in this process. Increased TGFβ1 expression is observed in the myocardium of human patients with idiopathic hypertrophic cardiomyopathy (Li et al. 1998). Consistently, TGFβ1 overexpression in transgenic mice results in hypertrophic growth of cardiomyocytes (Rosenkranz et al. 2002). Interestingly, this TGFβ1-induced cardiac hypertrophy is associated with increased myocardial β-adrenergic receptors (ARs) density (Rosenkranz et al. 2002) and β-AR blockade treatment in TGFβ1 transgenic mice prevents cardiac hypertrophy. On the other hand, angiotensin II was shown to induce TGFβ1 expression in myocardium and TGFβ1 (Wenzel et al. 2001) is required for AngII-mediated cardiacmocyte hypertrophy (Gray et al. 1998). However, the AT1 receptor blockade is insufficient to prevent the hypertrophic response in TGFβ transgenic mice (Rosenkranz et al. 2003), and TGFβ1 knockout mice are resistant to AngII-induced cardiac hypertrophy (Schultz Jel et al. 2002). Together, these data show that TGFβ1 plays a key role in AngII-mediated growth responses of cardiomyocytes via β-AR.
TGFβ1 and post-infarction inflammatory response
Cardiomyocyte death and hypoxia following infarction initiate inflammatory response, which leads to the infiltration of immune cells into the infarcted area, an important process for clearing debris from the wound and tissue repair (Mehta and Li 1999). In the meantime, a timely repression of inflammatory mediator synthesis is important for scar maturation. TGFβ1 plays a highly important and complex role in inflammatory response following cardiac injury (Celada and Maki 1992), It acts as a direct chemoattractant to monocytes (Wahl et al. 1987) and neutrophils (Fava et al. 1991) to recruit them to the infarct site. However, its effects on macrophages are primarily inhibitory (Frangogiannis et al. 2001). There is a need for a better understanding of the TGFβ1-modulated post-infarction inflammatory response for specific intervention that could attenuate inflammatory injury without interfering with myocardial healing.
TGFβ1 and cardiac neovascularisation
Genetic studies in mouse and human revealed that proper TGFβ1 signalling is essential for blood vessel formation (Chang et al. 2001; Harradine and Akhurst 2006). The vascular response to TGFβ1 is highly context-dependent and is shaped by factors such as ligand bioavailability and concentration, receptor availability and internalization, cross-talk with other signalling pathways, micro/marcovessel origin of vascular cells, and cellular density (Massague 2012). Although not fully understood, it is generally considered that TGFβ1 signalling in ECs occurs through TGFβRII recruiting either the ubiquitously expressed ALK5 or the EC-specific ALK1 (possibly with ALK5). Signalling via ALK5 leads to the activation of downstream transcription factors Smad 2 and 3, and thus plays an essential role in maintaining the vasculature at quiescent state. The TGFβ1/ALK1 signalling activates Smad 1, 5 and 8, resulting in increased EC migration, proliferation and angiogenesis (Goumans et al. 2003). The two pathways interact with each other at both receptor and the Smad level (Goumans et al. 2003), with ENG as a key player in switching TGFβ signalling toward the pro-angiogenic pathway (Lebrin et al. 2004). This paper showed that ENG regulates the balance of TGFβ signalling in endothelial cells by promoting TGFβ/ALK1 and inhibiting TGFβ/ALK5 signalling, and the subsequence endothelial proliferation.
Besides ECs, blood vessels comprise another essential component. The vascular mural cells support endothelium and are involved in blood vessel maturation and homeostasis. Similar to that in ECs, TGFβ1 mediates the proliferation of VSMC in a dose-dependent manner with high-dose being inhibitory and low-dose being stimulatory (Seay et al. 2005; Tsai et al. 2009). TGFβ1 induces the contractile phenotype of VSMCs by promoting the expression of α-smooth muscle actin and smooth muscle myosin (Hautmann et al. 1997; Seay et al. 2005). Perturbed TGFβ1 signalling results in failure of VSMC recruitment (Pardali et al. 2010) and the formation of aneurysm (Choudhary et al. 2009). TGFβ1 also stimulates the expression of plasminogen activator inhibitor (PAI)-1, a potent inhibitor of matrix MMPs, and therefore promotes blood vessel maturation by preventing the degradation of provisional matrix surrounding the nascent vessel (Jain 2003). On the other hand, the juxtaposition and collaboration between mural cells and ECs are important for local activation of latent TGFβ1, which further defines its context-dependent signalling in vascular cells (Antonelliorlidge et al. 1989; Sato et al. 1990).
Together, extensive investigations have shown that TGFβ1 modulates the signalling and function of different vascular cells and participates in multiple stages of blood vessel development, which makes it an attractive target for therapeutic angiogenesis. Indeed, it has been reported that exogenous application of TGFβ1 stimulates blood vessel formation in peripheral circulation (van Royen et al. 2002). This study showed that exogenous TGFβ1 promotes peripheral collateral artery formation and collateral circulation in rabbit hind limb model of femoral artery occlusion, partly by increasing monocyte adhesion and transmigration and enhancing the expression of growth factors and cytokines. Further studies are required to evaluate the impact of TGFβ treatment on functional blood vessel formation in the heart.
TGFβ1 signalling as a therapeutic target for heart failure?
TGFβ1 coordinates a broad spectrum of cellular processes that contributes to cardiac remodelling after MI and subsequent progression to heart failure (Bujak and Frangogiannis 2007). It can be beneficial or deleterious depending on the stage of disease development. For example, TGFβ1 plays a pivotal role in wound repair after infarction by suppressing inflammation, promoting the myofibroblast transition, and inducing blood vessel remodelling (Dobaczewski et al. 2011). However, prolonged TGFβ activation leads to excessive ECM deposition remote from the infarct site causing further damage to normal tissue architecture and cardiac function (Bujak et al. 2007). Indeed, inhibition of TGFβ before or immediately following MI led to further deterioration on cardiac function and increase mortality (Ikeuchi et al. 2004; Frantz et al. 2008), whereas its inhibition at 24 h post-MI attenuated remodelling with improved cardiac function in animal models of ischemic heart failure (Ikeuchi et al. 2004; Okada et al. 2005; Ellmers et al. 2008). In addition, due to its multifunctional and context dependent actions, a complete blockage of TGFβ signalling may cause undesirable side effects on immune regulation (Sasaki et al. 1992), angiogenesis (Bertolino et al. 2005), cancer surveillance (Salomon 2014) and wound healing (Faler et al. 2006). Taken together, TGFβ-targeted treatment must be carefully designed. A strategy that selectively attenuates the fibrogenic effect but stimulates the pro-angiogenic aspect of TGFβ1 may serve as an ideal treatment option for heart failure.
The interaction between LRG1 and TGFβ signalling
Leucine-rich α-2 glycoprotein 1 (LRG1) is a member of leucine-rich repeat (LRR) family of proteins, many of which are involved in protein–protein interactions, signalling and cell adhesion (Ng et al. 2011). Studies have shown that differential expression of LRG1 is associated with different types of cancer (Kawakami et al. 2005; Kakisaka et al. 2007; Ferrero et al. 2009; Andersen et al. 2010; Guergova-Kuras et al. 2011; Li et al. 2011; Sandanayake et al. 2011; Ladd et al. 2012; Linden et al. 2012, 2013; Liu et al. 2012; Wu et al. 2013; He et al. 2014; Wen et al. 2014), neurodegenerative disease (Miyajima et al. 2013), inflammatory diseases (Kentsis et al. 2012; Kharbanda et al. 2012; Serada et al. 2012), hydrocephalus (Li et al. 2006, 2007; Nakajima et al. 2010, 2011), heart failure (Watson et al. 2011), autoimmune disease (Serada et al. 2010), and ageing (Nakajima et al. 2012)
We found recently that LRG1 is expressed in quiescent vasculature at low levels but is significantly up-regulated together with TGFβ1 in remodelled and neovascular vessels in the eye (Wang et al. 2013). We showed that LRG1 interacts with multiple TGFβ receptors, especially ENG, which together with TGFβ1 further promotes the ability of LRG1 to bind angiogenic ALK1 but inhibits the association between LRG1 and angiostatic ALK5. The recruitment of LRG1 into the pro-angiogenic TGFβ receptor complex leads to enhanced Smad 1, 5 phosphorylation, EC proliferation, tube formation and blood vessel outgrowth. LRG1 inhibition by genetic knockout, siRNA knockdown or neutralizing antibodies led to reduced angiogenesis. In summary, our study showed that LRG1 plays a critical role in defining the context-dependent TGFβ signalling in ECs (Wang et al. 2013).
There is evidence that LRG1 is involved in other TGFβ-regulated processes. TGFβ is known to stimulate the expression of endothelin (ET1) (Ahmedat et al. 2012), an important molecule involved in myocardial hypertrophy and fibrosis. ET1 has been shown to inhibit LRG1 expression in dermal fibroblasts suggesting a potential role of LRG1 in TGFβ-mediated fibrosis (GEO accession GDS1980 / 1417290_at / Lrg1) (Vallender and Lahn 2006).
The involvement of LRG1 in cardiac remodelling and heart failure
LRG1 and ageing heart
As an unavoidable process of life, different systems of the body undergo progressive structural and functional alterations, and the heart is not an exception. With ageing, there is increased plaque formation in coronary arteries, cardiac wall thickness and interstitial fibrosis (Mendes et al. 2012; Dayal et al. 2013). These structural changes of myocardium are accompanied with concurrent vascular abnormalities, such as reduced diameter and density of collateral vessels, decreased vasodilation, and increased stiffness of vessel walls (Heil and Schaper 2004; Faber et al. 2011; Wang et al. 2011). In addition, there is a decreased expression and availability of growth factors such as hypoxia-inducible factor 1α and VEGF in response to hypoxic stress (Rivard et al. 2000), and ECs become less responsive to the stimulation of angiogenic growth factors with ageing (Lahteenvuo and Rosenzweig 2012). These alterations contribute to compromised cardiac function, increased susceptibility to damage and reduced ability to repair, which subsequently lead to increased incidence of heart failure in ageing population. Two separate studies have reported an decreased expression of LRG1 in the heart of aged mice compared to that in young mice (GDS2996 / 1417290_at / Lrg1 and GDS2972 / 97420_at / Lrg1) (Reiter et al. 2007). However, it is not clear whether the reduced LRG1 expression is the cause or the consequence of ageing-related structural and functional changes in the heart. Further studies are required to explore the role of LRG1 in specific patho-physiologies of the ageing heart and to discover whether it is possible to prevent or reverse age-dependent deterioration of the heart by overexpressing LRG1.
LRG1 and cardiac hypertrophy
Cardiac hypertrophy is the thickening of the heart wall in response to increased pressure or volume stress. Under certain conditions, such as during pregnancy or after sustained exercise, the enlargement of heart muscle is beneficial and is normally associated with a proportional increase in chamber dimensions and neovascularization (Catalucci et al. 2008). There is no concurrent fibrosis or reactivation of a foetal gene program in this physiological adaptation process (Beisvag et al. 2009). In addition, physiological hypertrophy does not cause increased risk of arrhythmia, impairment in cardiac function or future heart failure. Instead, exercise training has been shown to protect the heart against ageing-induced up-regulation of collagen deposition, collagen cross-linking, TIMP synthesis and down-regulation of active MMPs (Thomas et al. 2000, 2001; Kwak et al. 2008). The ageing-associated increase in extra-myocyte space was also significantly attenuated in rats which underwent exercise training (Kwak et al. 2008, 2011). Pathological cardiac hypertrophy, on the other hand, occurs as a consequence of hypertension, aortic stenosis, or other disease-causing stimuli. It is associated with significant structural abnormalities, which can lead to contractile dysfunction, arrhythmias and eventually heart failure (Scheuer et al. 1982; Breisch et al. 1986).
The activation of insulin-like growth factor-1 (IGF-1)/phosphoinositide 3-kinase (PI3K)/Akt pathway has been implicated in adaptive cardiac hypertrophy with endurance exercise (Neri Serneri et al. 2001). Studies have shown that transient activation of Akt1 leads to reversible cardiac hypertrophy, which is associated with reduced expression of Lrg1 (GDS2304 / 1417290_at / Lrg1) (Schiekofer et al. 2006). Consistent with this observation, a dominant negative form of PI3K significantly attenuates cardiac hypertrophy in the heart of transgenic mice overexpressing IGF1 receptor, which is also associated with a concurrent up-regulation of Lrg1 (GDS648 / 97420_at / Lrg1) (McMullen et al. 2004). However, a separate study showed no change in Lrg1 expression in transgenic mice with either constitutive active PI3K or dominant negative PI3K (GDS446 / 97420_at / Lrg1). Also, no change in Lrg1 expression was detected in the heart of rats following moderate physical training (GDS3134/1374626_at/Lrg1) (Giusti et al. 2009). These seemingly contradictory observations might be due to differences in the design of transgenic strategies, spatial and temporal expression pattern of transgenes, and the use of different animal species. Further studies are required to elucidate the role of LRG1 in physiological cardiac hypertrophy.
A reduced expression of LRG1 is observed in mouse models of pathological cardiac hypertrophy. LRG1 expression is significantly attenuated in the heart of mice with compensated pressure overload hypertrophy induced by transverse aortic constriction (TAC) (GDS794 / 97420_at / Lrg1, GDS3465 / 1417290_at / Lrg1) (Zhao et al. 2004; Smeets et al. 2008). A missense E180G mutation in α-tropomyosin (TM), an important contractile protein involved in sarcomeric function, is associated with familial hypertrophic cardiomyopathy (Chang et al. 2005). Transgenic mice overexpressing α-TM E180G exhibit severe cardiac hypertrophy characterized by myocyte disarray, asymmetric ventricular enlargement, fibrosis, cardiac arrhythmia and eventually die of heart failure (Michele et al. 2002). The expression of Lrg1 is significantly down-regulated in the ventricle of α-TM E180G transgenic mice (GDS2134 / 1417290_at / Lrg1) (Rajan et al. 2006).
Together, the literature supports lower LRG1 expression being associated with cardiac hypertrophy. Further studies are required to investigate the underlying molecular mechanism. Analysis of cardiac phenotypes in Lrg1−/− with exercise or pressure overload-induced hypertrophy will provide valuable information regarding the role of LRG1 in physiological and pathological cardiac hypertrophy. LRG1 overexpression might be able to reverse cardiac hypertrophy induced by the activation of IGF1/PI3K/Akt1 pathway, exercise, pressure overload, and in α-TM E180G transgenic mice. The activation of latent TGFβ requires functional and physical cooperation of mannose-6-phosphate (M6P)/IGF II receptor (IGFIIR) and the urokinase-type plasminogen activator receptor (uPAR) (Leksa et al. 2005). Furthermore, both TGFβ1 and IGF1 signal through the PI3K/Akt pathway and there is an extensive crosstalk between the two signalling pathways during cardiac fibrosis (Butt et al. 1995), cardiomyocyte apoptosis (Hynes et al. 2009), cardiac remodelling following myocardial infarction (Stavropoulou et al. 2010) and cardiac hypertrophy (Lisa et al. 2011). Understanding the role of LRG1 in TGFβ1 and IGF1 interactions may shed new light on the molecular mechanism of cardiac hypertrophy.
LRG1 and dilated cardiomyopathy
Muscle LIM protein (MLP) is a muscle-restricted cytoskeletal binding protein. The down-regulation of MLP protein is observed in human patients with idiopathic-dilated cardiomyopathy (Zolk et al. 2000). Consistent with this observation, MLP−/− mice develop dilated cardiomyopathy and eventually heart failure (Arber et al. 1997). Calsequestrin (CSQ) is a high-capacity sarcoplasmic reticulum Ca2+ binding protein. The myocardial-targeted overexpression of CSQ also leads to heart failure associated with dilated cardiomyopathy and left ventricular dysfunction (Jones et al. 1998). Both mouse models exhibited many key features present in the failing heart in human, such as functional β-AR uncoupling (Rockman et al. 1998). Advanced heart failure is normally developed in 6-month old MLP−/− mice and 14-week old CSQ transgenic mice. Despite different aetiologies, a decreased Lrg1 expression is associated with deterioration of cardiac function with lowest Lrg1 expression observed at the advanced stage of heart failure in both mouse models (GDS411 / aa172851_s_at / Lrg1) (Blaxall et al. 2003). However, the molecular basis of LRG1 down-regulation in both mouse models remains to be resolved, which is vital for dissecting the mechanism of dilated cardiomyopathy pathogenesis. LRG1 overexpression in myocardium might restore cardiac function and slow down the progression of heart failure in both models. Information extracted from this study will assist the designing effective treatment for dilated cardiomyopathy associated heart failure.
LRG1 and hypertension
Hypertension is a major risk factor for heart failure. To work against the high pressure, the heart must pump harder, which may lead to left ventricular hypertrophy and heart failure over time. The S.LWEx12x2x3x5 congenic rat is a model for hypertension and exhibits concentric cardiac hypertrophy. The expression of LRG1 is significantly down-regulated in the left ventricle of hypertensive S.LWEx12x2x3x5 rat exhibiting concentric cardiac hypertrophy with augmented contractile function (GDS3873 _ 1374626_at _ Lrg1) (Gopalakrishnan et al. 2011). However, it is not clear whether hypertension has a direct impact on the expression of Lrg1 or mediates LRG1 expression indirectly via hypertension-induced compensation. To study the expression of Lrg1 in other hypertension animal models with or without myocardial abnormalities will enrich our understanding of direct association between LRG1 and hypertension aetiology and development. LRG1 overexpression at the right dosage and right timing might prevent or reverse hypertension and hypertension-induced cardiomyopathy.
Perspectives
There is compelling evidence to suggest that TGFβ1 is associated with various cardiac pathologies involved in heart failure. Targeting TGFβ1 therefore represents an attractive strategy in managing progression of the disease. A number of therapeutic approaches for blocking the actions of TGFβ1 have been suggested, such as TGFβ1 neutralizing antibody (Kuwahara et al. 2002), soluble TGFβ receptor II (Okada et al. 2005), and small molecule inhibitors (Engebretsen et al. 2014). Some of these successfully attenuated cardiac fibrosis, decreased ventricular chamber dilation, improved cardiac function, and reduced mortality after infarction in preclinical studies (Kuwahara et al. 2002; Ellmers et al. 2008; Lian et al. 2010). However, given its role in angiogenesis, targeting TGFβ may affect collateral and microvessel formation, remodelling and perfusion following MI and cause increased burden of ischemic tissue. In addition, TGFβ1 is a pleiotropic cytokine with vital homeostatic functions. Broad TGFβ inhibition is likely to have adverse side effects, such as the development of autoimmune diseases, delayed wound healing, and tumour formation. Selectively targeting specific disease-driving aspects of TGFβ signalling at the right dose and timing and for an appropriate period is therefore critical in producing desirable therapeutic effects.
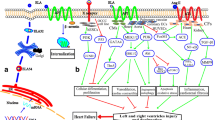
Our recent study led to the identification of a novel angiogenic factor, LRG1. In addition to ECs, LRG1 is also expressed in cardiac fibroblasts (Ifkovits et al. 2014) and cardiomyocytes (Chen et al. 2004). It is likely that LRG1 mediates TGFβ signalling through the ubiquitously expressed type I TGFβ receptor, ALK5, in non-ECs. There is convincing evidence that a decreased expression of LRG1 is associated with increased fibrosis, aberrant vascular properties, and altered cardiomyocytes characteristics in ageing heart, and in failing heart induced by genetic modification, pressure overload, or hypertension (Fig. 2). Understanding the molecular mechanism underlying the role of LRG1 and its regulation in cardiac remodelling process will assist the development of novel treatments for heart failure.

Potential role of LRG1 in cardiac remodelling. In the failing heart, the ability of blood vessels to respond to angiogenic factors is compromised, fibroblasts acquire myofibroblast phenotype by expressing increased ECM protein, and cardiomycytes are enlarged and undergo increased apoptosis. TGFβ1 mainly signals through ALK5 in ECs, cardiomyocyte and myofibroblasts leading to cardiac remodelling. In LRG1-treated heart, LRG1 switches TGFβ1 signalling towards the proangiogenic ALK1 signalling in ECs and promotes blood vessel formation. In myofibroblasts, LRG1 competes with TGFβ to bind ALK5 and antagonise TGFβ-induced ECM synthesis and to prevent fibrosis. With the presence of LRG1, there is reduced cardiomyocyte apoptosis and size
LRG1, a potential target for therapeutic angiogenesis
LRG1 promotes EC proliferation, tube formation and vessel outgrowth through regulating the endothelial TGFβ signalling (Wang et al. 2013). As it binds to the ubiquitously expressed type I TGFβ receptor, ALK5, LRG1 might mediate the signalling and function of other types of vascular cells and regulate blood vessel remodelling. TGFβ1 interacts with VEGF signalling at the receptor level (Glinka et al. 2011) and regulates VEGF expression in macrophages (Jeon et al. 2007) and ECs (Ferrari et al. 2006). Understanding the role of LRG1 in TGFβ1 and VEGF crosstalk may provide valuable information regarding the molecular mechanism of angiogenesis, and facilitate the development of novel therapeutic angiogenesis strategies. Angiogenic factors are normally released in a tightly controlled and timely manner. Lessons learned from previous studies suggested that the hypoxia condition of the ischemic tissue during treatment dramatically affects the benefit of therapeutic angiogenesis. Virus-based transgene delivery systems offer an opportunity for site-specific administration at the right location, time and dose. The route of delivery may also affect the efficacy of treatment. Direct delivery to the myocardium may be an effective method for therapeutic angiogenesis in the heart. Comorbidities are known to have a great impact on the outcome of therapeutic angiogenesis. It is therefore important to evaluate the impact of LRG1 on angiogenesis under heart failure-associated disease conditions such as diabetes, hypertension and obesity. As oedema can impose further burdens on ischemic tissue, studies are required to investigate the impact of LRG1 on blood permeability. It is worth noting that excessive angiogenesis contributes to cancer growth and metastasis, atherosclerotic plaque expansion and instability, arthritis and blinding eye diseases. Understanding the involvement of LRG1 in other vascular complications will help to predict potential side effects of the treatment. Taken together, LRG1 is an attractive target for therapeutic angiogenesis. Further studies are required to evaluate the efficacy and toxicity of LRG1 treatment for heart failure.
LRG1, a potential modulator of cardiac remodelling process
Cardiac fibrosis is a key contributor to the morbidity and mortality in heart failure. Despite intense research efforts, no effective treatment is available to control this detrimental process. Evidence showed that a reduced expression of LRG1 is associated with an increased TGFβ1 activity during the cardiac remodelling process in response to injury. However, it is not clear whether TGFβ1 exerts its function by inhibiting the expression of LRG1. As LRG1 binds ALK5 and TGFβRII independently of TGFβ1, it may exert its function by competing with TGFβ1 for binding with the ALK5/TGFβRII receptor complex. In addition to its role in mediating TGFβ1 signalling in endothelial cells, ENG plays an important role in cardiac fibrosis (Kapur et al. 2012). Unveiling the interaction between ENG and LRG1 in fibrosis will advance our understanding of the molecular mechanism underlying the cardiac remodelling process. It is widely accepted that context-dependent TGFβ1 signalling is defined by extensive interaction with other signalling pathways including IGF1 and AngII. Understanding the involvement of LRG1 in this highly complex network will shed light on the molecular mechanism of cardiac remodelling. Studies have shown that non-ECs including cardiac fibroblasts and cardiomyocytes also express LRG1. It will be interesting to see novel binding partners of LRG1 in these cells, and it will help to elucidate the TGFβ1-independent role of LRG1 in non-ECs in the heart. On the other hand, ECM is important for maintaining atherosclerotic plaque stability. Augment of collagen degradation has been correlated with ruptured plaques in patients (Cheng et al. 2009). Further studies are required to unveil the molecular and cellular mechanism of LRG1 in atherosclerotic plaque development and progression, which will provide valuable insights into the potential side effects of LRG1-targeted treatment.
Summary
At the moment, the focus of heart failure treatment is directed at risk factor management and alleviating symptoms; however, not all patients are suitable or will benefit from current therapeutics. As the magnitude of heart failure continues to accelerate globally, there is a pressing need for new treatments. Direct intervention on structural abnormalities of myocardium is a promising strategy. A reduced expression of LRG1 is associated with cardiac remodelling characterised by hypertrophy, fibrosis, and abnormal vasculature in various conditions leading to heart failure. However, it is not clear whether altered LRG1 expression is the cause or the consequence of these detrimental processes, or how LRG1 is correlated with specific pathologies and evolution of the disease. Although a great deal of work is still needed to fully understand the underlying mechanisms, targeting LRG1 and its regulators might offer a unique approach to treating heart failure by simultaneously targeting different pathologies of the disease.
References
Ahmedat AS et al (2012) beta(2)-adrenoceptors and muscarinic receptors mediate opposing effects on endothelin-1 expression in human lung fibroblasts. Eur J Pharmacol 691(1–3):218–224
Andersen JD, et al. (2010) Leucine-rich alpha-2-glycoprotein-1 is upregulated in sera and tumors of ovarian cancer patients. J Ovarian Res 3: 21
Antonelliorlidge A et al (1989) An activated form of transforming growth factor-beta is produced by cocultures of endothelial-cells and pericytes. Proc Natl Acad Sci U S A 86(12):4544–4548
Antoniucci D et al (2002) Relation of time to treatment and mortality in patients with acute myocardial infarction undergoing primary coronary angioplasty. Am J Cardiol 89(11):1248–1252
Arber S et al (1997) MLP-deficient mice exhibit a disruption of cardiac cytoarchitectural organization, dilated cardiomyopathy, and heart failure. Cell 88(3):393–403
Banerjee I et al (2006) Dynamic interactions between myocytes, fibroblasts, and extracellular matrix. Ann N Y Acad Sci 1080:76–84
Beisvag V et al (2009) Pathological and physiological hypertrophies are regulated by distinct gene programs. Eur J Cardiovasc Prev Rehabil 16(6):690–697
Bertolino P et al (2005) Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest 128(6 Suppl):585S–590S
Biernacka A et al (2011) TGF-beta signaling in fibrosis. Growth Factors 29(5):196–202
Blaxall BC et al (2003) Differential myocardial gene expression in the development and rescue of murine heart failure. Physiol Genomics 15(2):105–114
Breisch EA et al (1986) Exercise-induced cardiac hypertrophy: a correlation of blood flow and microvasculature. J Appl Physiol (1985) 60(4):1259–1267
Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74(2):184–195
Bujak M et al (2007) Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation 116(19):2127–2138
Butt RP et al (1995) Collagen production and replication by cardiac fibroblasts is enhanced in response to diverse classes of growth-factors. Eur J Cell Biol 68(3):330–335
Cambien F et al (1996) Polymorphisms of the transforming growth factor-beta 1 gene in relation to myocardial infarction and blood pressure. The Etude Cas-Temoin de l’Infarctus du Myocarde (ECTIM) Study. Hypertension 28(5):881–887
Cao Y (2009) Monotherapy versus combination therapy of angiogenic and arteriogenic factors for the treatment of ischemic disorders. Curr Mol Med 9(8):967–972
Cao Y et al (2005) Update on therapeutic neovascularization. Cardiovasc Res 65(3):639–648
Cao R et al (2010) VEGFR1-mediated pericyte ablation links VEGF and PlGF to cancer-associated retinopathy. Proc Natl Acad Sci U S A 107(2):856–861
Carmeliet P, Jain RK (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473(7347):298–307
Catalucci D et al (2008) Physiological myocardial hypertrophy: how and why? Front Biosci 13:312–324
Celada A, Maki RA (1992) Transforming growth factor-beta enhances the M-CSF and GM-CSF-stimulated proliferation of macrophages. J Immunol 148(4):1102–1105
Chang H et al (2001) Studying TGF-beta superfamily signaling by knockouts and knockins. Mol Cell Endocrinol 180(1–2):39–46
Chang AN et al (2005) Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem 280(40):34343–34349
Chen MM et al (2000) CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol 32(10):1805–1819
Chen H et al (2004) Gene expression changes associated with fibronectin-induced cardiac myocyte hypertrophy. Physiol Genomics 18(3):273–283
Cheng C et al (2009) Activation of MMP8 and MMP13 by angiotensin II correlates to severe intra-plaque hemorrhages and collagen breakdown in atherosclerotic lesions with a vulnerable phenotype. Atherosclerosis 204(1):26–33
Choi JH et al (2013) Frequency of myocardial infarction and its relationship to angiographic collateral flow in territories supplied by chronically occluded coronary arteries. Circulation 127(6):703–709
Choudhary B et al (2009) Absence of TGFbeta signaling in embryonic vascular smooth muscle leads to reduced lysyl oxidase expression, impaired elastogenesis, and aneurysm. Genesis 47(2):115–121
Cohn JN et al (2000) Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol 35(3):569–582
Cuevas P et al (1991) Hypotensive activity of fibroblast growth factor. Science 254(5035):1208–1210
Dayal S et al (2013) Hydrogen peroxide promotes ageing-related platelet hyperactivation and thrombosis. Circulation 127(12):1308–1316
Dean RG et al (2005) Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem 53(10):1245–1256
Dennis PA, Rifkin DB (1991) Cellular activation of latent transforming growth factor beta requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc Natl Acad Sci U S A 88(2):580–584
Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425(6958):577–584
Dobaczewski M et al (2011) Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J Mol Cell Cardiol 51(4):600–606
Ellmers LJ et al (2008) Transforming growth factor-beta blockade down-regulates the renin-angiotensin system and modifies cardiac remodeling after myocardial infarction. Endocrinology 149(11):5828–5834
Engebretsen KV et al (2014) Attenuated development of cardiac fibrosis in left ventricular pressure overload by SM16, an orally active inhibitor of ALK5. J Mol Cell Cardiol 76C:148–157
Euler-Taimor G, Heger J (2006) The complex pattern of SMAD signaling in the cardiovascular system. Cardiovasc Res 69(1):15–25
Faber JE et al (2011) Ageing causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler Thromb Vasc Biol 31(8):1748–1756
Faler BJ et al (2006) Transforming growth factor-beta and wound healing. Perspect Vasc Surg Endovasc Ther 18(1):55–62
Fava RA et al (1991) Transforming growth factor beta 1 (TGF-beta 1) induced neutrophil recruitment to synovial tissues: implications for TGF-beta-driven synovial inflammation and hyperplasia. J Exp Med 173(5):1121–1132
Ferrari G et al (2006) VEGF, a prosurvival factor, acts in concert with TGF-beta 1 to induce endothelial cell apoptosis. Proc Natl Acad Sci U S A 103(46):17260–17265
Ferrero S et al (2009) Increased expression of one isoform of leucine-rich alpha-2-glycoprotein in peritoneal fluid of women with uterine leiomyomas. Arch Gynecol Obstet 279(3):365–371
Forouzanfar MH et al (2013) Prevalence of heart failure by cause in 21 regions: global burden of diseases, injuries and risk factors-2010 study. J Am Coll Cardiol 61(10):E786
Fortuin FD et al (2003) One-year follow-up of direct myocardial gene transfer of vascular endothelial growth factor-2 using naked plasmid deoxyribonucleic acid by way of thoracotomy in no-option patients. Am J Cardiol 92(4):436–439
Frangogiannis NG et al (2001) Induction and suppression of interferon-inducible protein (IP)-10 in reperfused myocardial infarcts may regulate angiogenesis. Faseb J 15(6):1428–1430
Frantz S et al (2008) Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol 103(5):485–492
Giusti B et al (2009) Gene expression profile of rat left ventricles reveals persisting changes following chronic mild exercise protocol: implications for cardioprotection. BMC Genomics 10:342
Glennon PE et al (1995) Cellular mechanisms of cardiac hypertrophy. Br Heart J 73(6):496–499
Glinka Y et al (2011) Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis 32(4):613–621
Gopalakrishnan K et al (2011) Augmented rififylin is a risk factor linked to aberrant cardiomyocyte function, short-QT interval and hypertension. Hypertension 57(4):764–771
Goumans MJ et al (2003) Controlling the angiogenic switch: a balance between two distinct TGF-b receptor signaling pathways. Trends Cardiovasc Med 13(7):301–307
Gray MO et al (1998) Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res 40(2):352–363
Grines CL et al (2002) Angiogenic Gene Therapy (AGENT) trial in patients with stable angina pectoris. Circulation 105(11):1291–1297
Grines CL et al (2003) A randomized, double-blind, placebo-controlled trial of Ad5FGF-4 gene therapy and its effect on myocardial perfusion in patients with stable angina. J Am Coll Cardiol 42(8):1339–1347
Guergova-Kuras M et al (2011) Discovery of lung cancer biomarkers by profiling the plasma proteome with monoclonal antibody libraries. Mol Cell Proteomics 10(12):M111 010298
Habib GB et al (1991) Influence of coronary collateral vessels on myocardial infarct size in humans. Results of phase I thrombolysis in myocardial infarction (TIMI) trial. The TIMI Investigators. Circulation 83(3):739–746
Hao J et al (1999) Elevation of expression of Smads 2, 3, and 4, decorin and TGF-beta in the chronic phase of myocardial infarct scar healing. J Mol Cell Cardiol 31(3):667–678
Harada K et al (1994) Basic fibroblast growth factor improves myocardial function in chronically ischemic porcine hearts. J Clin Invest 94(2):623–630
Hariawala MD et al (1996) VEGF improves myocardial blood flow but produces EDRF-mediated hypotension in porcine hearts. J Surg Res 63(1):77–82
Harradine KA, Akhurst RJ (2006) Mutations of TGF beta signaling molecules in human disease. Ann Med 38(6):403–414
Hautmann MB et al (1997) A transforming growth factor beta (TGFbeta) control element drives TGFbeta-induced stimulation of smooth muscle alpha-actin gene expression in concert with two CArG elements. J Biol Chem 272(16):10948–10956
He X et al (2014) Screening differential expression of serum proteins in AFP-negative HBV-related hepatocellular carcinoma using iTRAQ -MALDI-MS/MS. Neoplasma 61(1):17–26
Hedlund EM et al (2009) Malignant cell-derived PlGF promotes normalization and remodeling of the tumor vasculature. Proc Natl Acad Sci U S A 106(41):17505–17510
Heil M, Schaper W (2004) Influence of mechanical, cellular, and molecular factors on collateral artery growth (arteriogenesis). Circ Res 95(5):449–458
Heinl-Green A et al (2005) The efficacy of a ‘master switch gene’ HIF-1 alpha in a porcine model of chronic myocardial ischaemia. Eur Heart J 26(13):1327–1332
Hendel RC et al (2000) Effect of intracoronary recombinant human vascular endothelial growth factor on myocardial perfusion: evidence for a dose-dependent effect. Circulation 101(2):118–121
Henry TD et al (2001) Intracoronary administration of recombinant human vascular endothelial growth factor to patients with coronary artery disease. Am Heart J 142(5):872–880
Heuser R et al (2000) A retrospective study of 6,671 patients comparing coronary stenting and balloon angioplasty. J Invasive Cardiol 12(7):354–362
Ho CY et al (2010) Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med 363(6):552–563
Horowitz JR et al (1997) Vascular endothelial growth factor/vascular permeability factor produces nitric oxide-dependent hypotension. Evidence for a maintenance role in quiescent adult endothelium. Arterioscler Thromb Vasc Biol 17(11):2793–2800
Horvath KA (2000) Transmyocardial laser revascularization in the treatment of myocardial ischemia. J Card Surg 15(4):271–277
Hynes B et al (2009) Endothelial progenitor cell derived conditioned media reduces in vivo cardiomyocyte apoptosis acting through TGFB1 and IGF1. J Am Coll Cardiol 53(10):A313–A314
Ifkovits JL et al (2014) Inhibition of TGFbeta signaling increases direct conversion of fibroblasts to induced cardiomyocytes. PLoS ONE 9(2):e89678
Ikeuchi M et al (2004) Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res 64(3):526–535
Inoue M et al (1998) Vascular endothelial growth factor (VEGF) expression in human coronary atherosclerotic lesions: possible pathophysiological significance of VEGF in progression of atherosclerosis. Circulation 98(20):2108–2116
Iredale JP (2007) Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest 117(3):539–548
Jain RK (2003) Molecular regulation of vessel maturation. Nat Med 9(6):685–693
Jeon SH et al (2007) Mechanisms underlying TGF-beta 1-induced expression of VEGF and Flk-1 in mouse macrophages and their implications for angiogenesis. J Leukoc Biol 81(2):557–566
Johnson FL (2014) Pathophysiology and etiology of heart failure. Cardiol Clin 32(1):9–19, vii
Jones LR et al (1998) Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest 101(7):1385–1393
Kakisaka T et al (2007) Plasma proteomics of pancreatic cancer patients by multi-dimensional liquid chromatography and two-dimensional difference gel electrophoresis (2D-DIGE): up-regulation of leucine-rich alpha-2-glycoprotein in pancreatic cancer. J Chromatogr B 852(1–2):257–267
Kapur NK et al (2012) Reduced endoglin activity limits cardiac fibrosis and improves survival in heart failure. Circulation 125(22):2728–2738
Kapur NK et al (2013) Endoglin: a critical mediator of cardiovascular health. Vasc Health Risk Manag 9:195–206
Karagueuzian HS (2011) Targeting cardiac fibrosis: a new frontier in antiarrhythmic therapy? Am J Cardiovasc Dis 1(2):101–109
Kastrup J et al (2011) A randomised, double-blind, placebo-controlled, multicentre study of the safety and efficacy of BIOBYPASS (AdGVVEGF121.10NH) gene therapy in patients with refractory advanced coronary artery disease: the NOVA trial. EuroIntervention 6(7):813–818
Kaul U et al (1991) Restenosis after successful coronary angioplasty in single vessel disease. Indian Heart J 43(1):11–15
Kawakami T et al (2005) Proteomic analysis of sera from hepatocellular carcinoma patients after radiofrequency ablation treatment. Proteomics 5(16):4287–4295
Kehat I, Molkentin JD (2010) Molecular pathways underlying cardiac remodeling during pathophysiological stimulation. Circulation 122(25):2727–2735
Kentsis A et al (2012) Detection and diagnostic value of urine leucine-rich alpha-2-glycoprotein in children with suspected acute appendicitis. Ann Emerg Med 60(1):78–83
Khalil N (1999) TGF-beta: from latent to active. Microbes Infect 1:1255–1263
Khan R, Sheppard R (2006) Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology 118(1):10–24
Kharbanda AB et al (2012) Novel serum and urine markers for pediatric appendicitis. Acad Emerg Med 19(1):56–62
Kim I et al (2001) Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem 276(10):7614–7620
Koitabashi N et al (2011) Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121(6):2301–2312
Krum H et al (2002) Which factors mediate cardiac fibrosis following myocardial infarction? Role of TGF beta 1 & CTGF in pathological collagen deposition post-MI. Circulation 106(19):232
Kuwahara F et al (2002) Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 106(1):130–135
Kwak HB et al (2008) Exercise training attenuates extracellular matrix remodeling in the ageing Rat heart. Med Sci Sports Exerc 40(5):S459
Kwak HB et al (2011) Exercise training reduces fibrosis and matrix metalloproteinase dysregulation in the ageing rat heart. Faseb J 25(3):1106–1117
Ladd JJ et al (2012) Increased plasma levels of the APC-interacting protein MAPRE1, LRG1, and IGFBP2 preceding a diagnosis of colorectal cancer in women. Cancer Prev Res (Phila) 5(4):655–664
Lahteenvuo J, Rosenzweig A (2012) Effects of ageing on angiogenesis. Circ Res 110(9):1252–1264
Landau C et al (1995) Intrapericardial basic fibroblast growth-factor induces myocardial angiogenesis in a rabbit model of chronic ischemia. Am Heart J 129(5):924–931
Lazarous DF et al (1996) Comparative effects of basic fibroblast growth factor and vascular endothelial growth factor on coronary collateral development and the arterial response to injury. Circulation 94(5):1074–1082
Leask A (2007) TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc Res 74(2):207–212
Lebrin F et al (2004) Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J 23(20):4018–4028
Lee LY et al (2000) Focal angiogen therapy using intramyocardial delivery of an adenovirus vector coding for vascular endothelial growth factor 121. Ann Thorac Surg 69(1):14–23
Leksa V et al (2005) TGF-beta-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J Cell Sci 118(Pt 19):4577–4586
Li G et al (1998) Elevated insulin-like growth factor-I and transforming growth factor-beta 1 and their receptors in patients with idiopathic hypertrophic obstructive cardiomyopathy. A possible mechanism. Circulation 98(19 Suppl):II144–II149. discussion II149-150
Li X et al (2006) Analysis of potential diagnostic biomarkers in cerebrospinal fluid of idiopathic normal pressure hydrocephalus by proteomics. Acta Neurochir (Wien) 148(8):859–864. discussion 864
Li X et al (2007) Expression of TGF-betas and TGF-beta type II receptor in cerebrospinal fluid of patients with idiopathic normal pressure hydrocephalus. Neurosci Lett 413(2):141–144
Li Y et al (2011) Proteomic identification of exosomal LRG1: a potential urinary biomarker for detecting NSCLC. Electrophoresis 32(15):1976–1983
Lian RQ et al (2010) Soluble transforming growth factor-beta 1 receptor II might inhibit transforming growth factor-beta-induced myofibroblast differentiation and improve ischemic cardiac function after myocardial infarction in rats. Coron Artery Dis 21(6):369–377
Linden M et al (2012) Proteomic analysis of urinary biomarker candidates for nonmuscle invasive bladder cancer. Proteomics 12(1):135–144
Linden M et al (2013) Tumour expression of bladder cancer-associated urinary proteins. BJU Int 112(3):407–415
Lisa M et al (2011) Insulin-Like Growth Factor-1 (IGF-1) Reduces ischemic changes and increases circulating angiogenic factors in experimentally - induced myocardial infarction in rats. Vasc Cell 3(1):13
Liu YS et al (2012) Shotgun and targeted proteomics reveal that pre-surgery serum levels of LRG1, SAA, and C4BP may refine prognosis of resected squamous cell lung cancer. J Mol Cell Biol 4(5):344–347
Lopez JJ et al (1998) Angiogenic potential of perivascularly delivered aFGF in a porcine model of chronic myocardial ischemia. Am J Physiol 274(3 Pt 2):H930–H936
Lopez B et al (2001) Biochemical assessment of myocardial fibrosis in hypertensive heart disease. Hypertension 38(5):1222–1226
Losordo DW et al (1998) Gene therapy for myocardial angiogenesis - Initial clinical results with direct myocardial injection of phVEGF(165) as sole therapy for myocardial ischemia. Circulation 98(25):2800–2804
Losordo DW et al (2002) Phase 1/2 placebo-controlled, double-blind, dose-escalating trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation 105(17):2012–2018
Manabe I et al (2002) Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res 91(12):1103–1113
Mann DL, Chakinala M (2012) Heart failure and Cor Pulmonale. Chapter 234 In: Harrison's Internal Medicine. McGraw-Hill, New York
Massague J (2012) TGFbeta signalling in context. Nat Rev Mol Cell Biol 13(10):616–630
McMullen JR et al (2004) Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol Cell Biol 24(14):6231–6240
Mehta JL, Li DY (1999) Inflammation in ischemic heart disease: response to tissue injury or a pathogenetic villain? Cardiovasc Res 43(2):291–299
Meier P et al (2012) The impact of the coronary collateral circulation on mortality: a meta-analysis. Eur Heart J 33(5):614–621
Mendes AB et al (2012) Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Med (B Aires) 72(3):216–220
Michele DE et al (2002) Cardiac dysfunction in hypertrophic cardiomyopathy mutant tropomyosin mice is transgene-dependent, hypertrophy-independent, and improved by beta-blockade. Circ Res 91(3):255–262
Millan FA et al (1991) Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development 111(1):131–143
Miyajima M et al. (2013). Leucine-Rich alpha 2-Glycoprotein Is a Novel Biomarker of Neurodegenerative Disease in Human Cerebrospinal Fluid and Causes Neurodegeneration in Mouse Cerebral Cortex. PLoS ONE 8(9):e74453
Nagy JA et al (2007) VEGF-A and the induction of pathological angiogenesis. Annu Rev Pathol 2:251–275
Nakajima M et al (2010) Diagnostic value of CSF biomarker profile in idiopathic normal pressure hydrocephalus; leucine-rich alpha-2-glycoprotein is a potential biological marker. Rinsho Shinkeigaku 50(11):973–976
Nakajima M et al (2011) Leucine-rich alpha-2-glycoprotein is a marker for idiopathic normal pressure hydrocephalus. Acta Neurochir 153(6):1339–1346
Nakajima M et al (2012) Brain localization of leucine-rich alpha 2-glycoprotein and its role. Acta Neurochir Suppl 113:97–101
Neri Serneri G, Boddi M, Modesti P, Cecioni I, Coppo M, Padeletti L, Michelucci A, Colella A, Galanti G (2001) Increased cardiac sympathetic activity and insulin-like growth factor-I formation are associated with physiological hypertrophy in athletes. Circ Res 89:977–982
Ng AC et al (2011) Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci U S A 108(Suppl 1):4631–4638
Nishimura RA et al (2014) 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the american college of cardiology/american heart association task force on practice guidelines. Circulation 129(23):e521–e643
Norgren L et al (2007) Inter-society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg 45(Suppl S):S5–S67
Okada H et al (2005) Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation 111(19):2430–2437
Pardali E et al (2010) Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends Cell Biol 20(9):556–567
Pauschinger M et al (1999) Dilated cardiomyopathy is associated with significant changes in collagen type I/III ratio. Circulation 99(21):2750–2756
Qiao B et al (2005) Transforming growth factor (TGF)-beta-activated kinase 1 mimics and mediates TGF-beta-induced stimulation of type II collagen synthesis in chondrocytes independent of Col2a1 transcription and Smad3 signaling. J Biol Chem 280(17):17562–17571
Rajan S et al (2006) Microarray analysis of gene expression during early stages of mild and severe cardiac hypertrophy. Physiol Genomics 27(3):309–317
Reilly JP et al (2005) Long-term (2-year) clinical events following transthoracic intramyocardial gene transfer of VEGF-2 in no-option patients. J Interv Cardiol 18(1):27–31
Reiter E et al (2007) Anti-inflammatory properties of alpha- and gamma-tocopherol. Mol Asp Med 28(5–6):668–691
Rivard A et al (2000) Age-dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. J Biol Chem 275(38):29643–29647
Rockman HA et al (1998) Expression of a beta-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A 95(12):7000–7005
Rosengart TK et al (1999) Angiogenesis gene therapy: phase I assessment of direct intramyocardial administration of an adenovirus vector expressing VEGF121 cDNA to individuals with clinically significant severe coronary artery disease. Circulation 100(5):468–474
Rosenkranz S et al (2002) Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol 283(3):H1253–H1262
Rosenkranz S et al (2003) beta-adrenoceptor blockade prevents cardiac hypertrophy and failure in transforming growth factor-beta 1 transgenic mice. Eur Heart J 24:665
Ross R (1993) Atherosclerosis: current understanding of mechanisms and future strategies in therapy. Transplant Proc 25(2):2041–2043
Salomon D (2014) Transforming growth factor beta in cancer: Janus, the two-faced god. J Natl Cancer Inst 106(2):djt441
Sandanayake NS et al (2011) A combination of serum leucine-rich alpha-2-glycoprotein 1, CA19-9 and interleukin-6 differentiate biliary tract cancer from benign biliary strictures. Br J Cancer 105(9):1370–1378
Sanderson JE et al (2001) Transforming growth factor-beta(1) expression in dilated cardiomyopathy. Heart 86(6):701–708
Sasaki H et al (1992) Transforming growth factor-beta in the regulation of the immune response. Clin Immunol Immunopathol 65(1):1–9
Sato Y et al (1990) Characterization of the activation of latent TGF-beta by co-cultures of endothelial cells and pericytes or smooth muscle cells: a self-regulating system. J Cell Biol 111(2):757–763
Scheuer J et al (1982) Physiologic cardiac hypertrophy corrects contractile protein abnormalities associated with pathologic hypertrophy in rats. J Clin Invest 70(6):1300–1305
Schiekofer S et al (2006) Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure. Physiol Genomics 27(2):158–170
Schultz Jel J et al (2002) TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest 109(6):787–796
Schumacher B et al (1998) Induction of neoangiogenesis in ischemic myocardium by human growth factors: first clinical results of a new treatment of coronary heart disease. Circulation 97(7):645–650
Scott CD, Firth SM (2004) The role of the M6P/IGF-II receptor in cancer: Tumor suppression or garbage disposal? Horm Metab Res 36(5):261–271
Seay U et al (2005) Transforming growth factor-beta-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J Pharmacol Exp Ther 315(3):1005–1012
Seeland U et al (2002) Myocardial fibrosis in transforming growth factor-beta(1) (TGF-beta(1)) transgenic mice is associated with inhibition of interstitial collagenase. Eur J Clin Invest 32(5):295–303
Serada S et al (2010) iTRAQ-based proteomic identification of leucine-rich alpha-2 glycoprotein as a novel inflammatory biomarker in autoimmune diseases. Ann Rheum Dis 69(4):770–774
Serada S et al (2012) Serum leucine-rich alpha-2 glycoprotein is a disease activity biomarker in ulcerative colitis. Inflamm Bowel Dis 18(11):2169–2179
Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113(6):685–700
Shi ML et al (2011) Latent TGF-beta structure and activation. Nature 474(7351):343–U370
Shou M et al (1997) Effect of basic fibroblast growth factor on myocardial angiogenesis in dogs with mature collateral vessels. J Am Coll Cardiol 29(5):1102–1106
Simons M et al (2002) Pharmacological treatment of coronary artery disease with recombinant fibroblast growth factor-2: double-blind, randomized, controlled clinical trial. Circulation 105(7):788–793
Smeets PJ et al (2008) Transcriptomic analysis of PPARalpha-dependent alterations during cardiac hypertrophy. Physiol Genomics 36(1):15–23
Stavropoulou A et al (2010) uPA, uPAR and TGF beta(1) expression during early and late post myocardial infarction period in Rat myocardium. In Vivo 24(5):647–652
Symes JF et al (1999) Gene therapy with vascular endothelial growth factor for inoperable coronary artery disease. Ann Thorac Surg 68(3):830–836. discussion 836–837
Thannickal VJ et al (2003) Myofibroblast differentiation by transforming growth factor-beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem 278(14):12384–12389
Thomas DP et al (2000) Collagen gene expression in rat left ventricle: interactive effect of age and exercise training. J Appl Physiol 89(4):1462–1468
Thomas DP et al (2001) Exercise training attenuates ageing-associated increases in collagen and collagen crosslinking of the left but not the right ventricle in the rat. Eur J Appl Physiol 85(1–2):164–169
Thurston G (2002) Complementary actions of VEGF and angiopoietin-1 on blood vessel growth and leakage. J Anat 200(6):575–580
Tsai S et al (2009) TGF-beta through Smad3 signaling stimulates vascular smooth muscle cell proliferation and neointimal formation. Am J Physiol Heart Circ Physiol 297(2):H540–H549
Udelson JE et al (2000) Therapeutic angiogenesis with recombinant fibroblast growth factor-2 improves stress and rest myocardial perfusion abnormalities in patients with severe symptomatic chronic coronary artery disease. Circulation 102(14):1605–1610
Unger EF et al (1994) Basic fibroblast growth factor enhances myocardial collateral flow in a canine model. Am J Physiol 266(4 Pt 2):H1588–H1595
Unger EF et al (2000) Effects of a single intracoronary injection of basic fibroblast growth factor in stable angina pectoris. Am J Cardiol 85(12):1414–1419
Vale PR et al (2001) Randomized, single-blind, placebo-controlled pilot study of catheter-based myocardial gene transfer for therapeutic angiogenesis using left ventricular electromechanical mapping in patients with chronic myocardial ischemia. Circulation 103(17):2138–2143
Vallender TW, Lahn BT (2006) Localized methylation in the key regulator gene endothelin-1 is associated with cell type-specific transcriptional silencing. FEBS Lett 580(18):4560–4566
van Meeteren LA, ten Dijke P (2012) Regulation of endothelial cell plasticity by TGF-beta. Cell Tissue Res 347(1):177–186
van Royen N et al (2002) Exogenous application of transforming growth factor beta 1 stimulates arteriogenesis in the peripheral circulation. FASEB J 16(3):432–434
Villarreal FJ, Dillmann WH (1992) Cardiac hypertrophy-induced changes in mRNA levels for TGF-beta 1, fibronectin, and collagen. Am J Physiol 262(6 Pt 2):H1861–H1866
Wahl SM et al (1987) Transforming growth factor type beta induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci U S A 84(16):5788–5792
Wang JS et al (2011) Ageing-induced collateral dysfunction: impaired responsiveness of collaterals and susceptibility to apoptosis via dysfunctional eNOS signaling. J Cardiovasc Transl Res 4(6):779–789
Wang X et al (2013) LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. Nature 499(7458):306–311
Watson CJ et al (2011) Proteomic analysis of coronary sinus serum reveals leucine-rich alpha 2-glycoprotein as a novel biomarker of ventricular dysfunction and heart failure. Circ Heart Fail 4(2):188–U126
Weber KT et al (1995) Structural remodelling of the heart by fibrous tissue: role of circulating hormones and locally produced peptides. Eur Heart J 16:12–18
Wen SY et al (2014) LRG1 is an independent prognostic factor for endometrial carcinoma. Tumor Biol 35(7):7125–7133
Wenzel S et al (2001) Redox-sensitive intermediates mediate angiotensin II-induced p38 MAP kinase activation, AP-1 binding activity, and TGF-beta expression in adult ventricular cardiomyocytes. FASEB J 15(12):2291–2293
Wu J et al (2013) Altered expression of sialylated glycoproteins in ovarian cancer sera using lectin-based ELISA assay and quantitative glycoproteomics analysis. J Proteome Res 12(7):3342–3352
Zeisberg EM et al (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med 13(8):952–961
Zhang DZ et al (2002) Efficacy and safety of therapeutic angiogenesis from direct myocardial administration of an adenoviral vector expressing vascular endothelial growth factor 165. Chin Med J 115(5):643–648
Zhao M et al (2004) Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics 19(1):93–105
Zolk O et al (2000) Decreased expression of the cardiac LIM domain protein MLP in chronic human heart failure. Circulation 101(23):2674–2677
Compliance with Ethical Standards
Funding
This work is supported by Cooperative Basic Research Grant (CBRG) CBRG13nov from NMRC Singapore.
Conflict of interest
Weihua Song declares that he has no conflict of interest. Xiaomeng Wang declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: Biophysics of Human Heart Failure
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Song, W., Wang, X. The role of TGFβ1 and LRG1 in cardiac remodelling and heart failure. Biophys Rev 7, 91–104 (2015). https://doi.org/10.1007/s12551-014-0158-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-014-0158-y