
Abstract
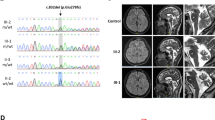
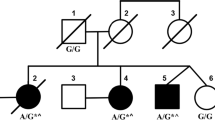
Spinocerebellar ataxia type 28 (SCA28) is an autosomal dominant neurodegenerative disorder caused by missense AFG3L2 mutations. To examine the occurrence of SCA28 in the Czech Republic, we screened 288 unrelated ataxic patients with hereditary (N = 49) and sporadic or unknown (N = 239) form of ataxia for mutations in exons 15 and 16, the AFG3L2 mutation hotspots. A single significant variant, frameshift mutation c.1958dupT leading to a premature termination codon, was identified in a patient with slowly progressive speech and gait problems starting at the age of 68 years. Neurological examination showed cerebellar ataxia, mild Parkinsonian features with predominant bradykinesia, polyneuropathy of the lower limbs, and cognitive decline. However, other common SCA28 features like pyramidal tract signs (lower limb hyperreflexia, positive Babinski sign), ophthalmoparesis or ptosis were absent. The mutation was also found in a patient’s unaffected daughter in whom a targeted examination at 53 years of age revealed mild imbalance signs. RNA analysis showed a decreased ratio of the transcript from the mutated AFG3L2 allele relative to the normal transcript in the peripheral lymphocytes of both patients. The ratio was increased by puromycin treatment, indicating that the mutated transcript can be degraded via nonsense-mediated RNA decay. The causal link between the mutation and the phenotype of the patient is currently unclear but a pathogenic mechanism based on AFG3L2 haploinsufficiency rather than the usual dominant-negative effect of missense AFG3L2 mutations reported in SCA28, cannot be excluded.


Similar content being viewed by others

References
Duarri A, Jezierska J, Fokkens M, Meijer M, Schelhaas HJ, den Dunnen WF, et al. Mutations in potassium channel kcnd3 cause spinocerebellar ataxia type 19. Ann Neurol. 2012;72:870–80.
Lee YC, Durr A, Majczenko K, Huang YH, Liu YC, Lien CC, et al. Mutations in KCND3 cause spinocerebellar ataxia type 22. Ann Neurol. 2012;72:859–69.
Matilla-Duenas A. The ever expanding spinocerebellar ataxias. Editorial. Cerebellum. 2012;11:821–7.
Cagnoli C, Mariotti C, Taroni F, Seri M, Brussino A, Michielotto C, et al. SCA28, a novel form of autosomal dominant cerebellar ataxia on chromosome 18p11.22-q11.2. Brain. 2006;129:235–42.
Di Bella D, Lazzaro F, Brusco A, Plumari M, Battaglia G, Pastore A, et al. Mutations in the mitochondrial protease gene AFG3L2 cause dominant hereditary ataxia SCA28. Nat Genet. 2010;42:313–21.
Mariotti C, Brusco A, Di Bella D, Cagnoli C, Seri M, Gellera C, et al. Spinocerebellar ataxia type 28: a novel autosomal dominant cerebellar ataxia characterized by slow progression and ophthalmoparesis. Cerebellum. 2008;7:184–8.
Cagnoli C, Stevanin G, Brussino A, Barberis M, Mancini C, Margolis RL, et al. Missense mutations in the AFG3L2 proteolytic domain account for approximately 1.5 % of European autosomal dominant cerebellar ataxias. Hum Mutat. 2010;31:1117–24.
Edener U, Wollner J, Hehr U, Kohl Z, Schilling S, Kreuz F, et al. Early onset and slow progression of SCA28, a rare dominant ataxia in a large four-generation family with a novel AFG3L2 mutation. Eur J Hum Genet. 2010;18:965–8.
Pierson TM, Adams D, Bonn F, Martinelli P, Cherukuri PF, Teer JK, et al. Whole-exome sequencing identifies homozygous AFG3L2 mutations in a spastic ataxia-neuropathy syndrome linked to mitochondrial m-AAA proteases. PLoS Genet. 2011;7:e1002325.
Banfi S, Bassi MT, Andolfi G, Marchitiello A, Zanotta S, Ballabio A, et al. Identification and characterization of AFG3L2, a novel paraplegin-related gene. Genomics. 1999;59:51–8.
Nolden M, Ehses S, Koppen M, Bernacchia A, Rugarli EI, Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–89.
Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. Embo J. 2008;27:306–14.
Koppen M, Metodiev MD, Casari G, Rugarli EI, Langer T. Variable and tissue-specific subunit composition of mitochondrial m-AAA protease complexes linked to hereditary spastic paraplegia. Mol Cell Biol. 2007;27:758–67.
Casari G, De Fusco M, Ciarmatori S, Zeviani M, Mora M, Fernandez P, et al. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–83.
Klebe S, Depienne C, Gerber S, Challe G, Anheim M, Charles P, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain. 2012;135:2980–93.
van Gassen KL, van der Heijden CD, de Bot ST, den Dunnen WF, van den Berg LH, Verschuuren-Bemelmans CC, et al. Genotype-phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain. 2012;135:2994–3004.
Almajan ER, Richter R, Paeger L, Martinelli P, Barth E, Decker T, et al. AFG3L2 supports mitochondrial protein synthesis and Purkinje cell survival. J Clin Invest. 2012;122:4048–58.
Maltecca F, De Stefani D, Cassina L, Consolato F, Wasilewski M, Scorrano L, et al. Respiratory dysfunction by AFG3L2 deficiency causes decreased mitochondrial calcium uptake via organellar network fragmentation. Hum Mol Genet. 2012;21:3858–70.
Hwang J, Kim YK. When a ribosome encounters a premature termination codon. BMB Rep. 2013;46:9–16.
Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–12.
Magyar I, Colman D, Arnold E, Baumgartner D, Bottani A, Fokstuen S, et al. Quantitative sequence analysis of FBN1 premature termination codons provides evidence for incomplete NMD in leukocytes. Hum Mutat. 2009;30:1355–64.
Maltecca F, Magnoni R, Cerri F, Cox GA, Quattrini A, Casari G. Haploinsufficiency of AFG3L2, the gene responsible for spinocerebellar ataxia type 28, causes mitochondria-mediated Purkinje cell dark degeneration. J Neurosci. 2009;29:9244–54.
Turleau C. Monosomy 18p. Orphanet J Rare Dis. 2008;3:4.
Kim JI, Ju YS, Park H, Kim S, Lee S, Yi JH, et al. A highly annotated whole-genome sequence of a Korean individual. Nature. 2009;460:1011–5.
Acknowledgments
We thank the patients for their participation in the study, two anonymous reviewers for very valuable comments and Michal Krupka, Ph.D., for his help with Western blot analysis. This work was supported by grant NT-12221 and grant for conceptual development of research organization University Hospital Motol 00064203 from the Ministry of Health of the Czech Republic, and by grant IGA-LF_13_13 from the Internal grant agency of Palacky University.
Conflict of interest
There are no potential conflicts of interest that might bias this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Musova, Z., Kaiserova, M., Kriegova, E. et al. A Novel Frameshift Mutation in the AFG3L2 Gene in a Patient with Spinocerebellar Ataxia. Cerebellum 13, 331–337 (2014). https://doi.org/10.1007/s12311-013-0538-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-013-0538-z