
Abstract
Major depressive disorder causes significant morbidity, affecting people’s ability to work, function in relationships, and engage in social activities. Moreover, major depressive disorder increases the risk of suicidal ideation, attempted suicide and death by completed suicide. There is evidence that chronic stress can cause major depressive disorder. As for genetic factors, only minor susceptibility genes have been reliably identified. The serotonin system provides a logical source of susceptibility genes for depression, because this system is the target of selective serotonin reuptake-inhibitor drugs that are effective in treating depression. The 5-hydroxytryptamine (serotonin) transporter (5-HTT) has received particular attention because it is involved in the reuptake of serotonin at brain synapses. One common polymorphic variant of the 5-HTT-linked polymorphic region (5-HTTLPR), which affects the promoter of the 5-HTT gene, causes reduced uptake of the neurotransmitter serotonin into the presynaptic cells in the brain. The authors discussed the relationship between genetic polymorphisms and major depressive disorder, with special emphasis on the 5-HTTTLPR polymorphism. As the 5-HTTLPR polymorphism was significantly associated with an increased risk of major depressive disorder, the 5-HTT gene may be a candidate for a major depressive disorder susceptibility gene. As major depressive disorder is a multifactorial disease, an improved understanding of the interplay of environmental and genetic polymorphisms at multiple loci may help identify individuals who are at increased risk for major depressive disorder. Hopefully, in the future we will be able to screen for major depressive disorder susceptibility by using specific biomarkers.
Similar content being viewed by others

Introduction
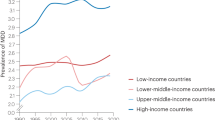
The World Health Organization (WHO) has estimated that each year about 877,000 people die from suicide. The WHO has further reported that suicide attempts are up to 20 times more frequent than completed suicides and that mental health disorders (particularly depression and substance abuse) are associated with more than 90% of all cases of suicide [1]. Depression causes significant morbidity, affecting people’s ability to work, function in relationships, and engage in social activities. Major depressive disorder increases the risk of suicidal ideation, attempted suicide and death by completed suicide. The lifetime of death by suicide in major depressive disorder defined by the Diagnostic and Statistical Manual of Mental Disorders fourth edition (DSM-IV) [2] is frequently cited as 15% [3]. The new generation of antidepressants cannot be used by depressed patients to kill themselves [4]. Therefore, the prevalence of death from suicide among today’s outpatients may include patients with other mental disorders, such as schizophrenia, and that among outpatients with depression may be overestimated. Nierenberg et al. [5] suggested that the actual lifetime risk of death from suicide in major depressive disorder was between 3.4 and 6%, but this estimate may omit important discrepancies among different ethnic populations. Culture can influence diagnosis, experience, and communication of symptoms. Some patients may have difficulty saying “I am sad” or “I am depressed,” but others can more easily say “I have bad nerves” or report vague somatic symptoms [6]. Simon et al. [7] examined the relationship between somatic symptoms and depression in general health care. After screening about 26,000 patients in 14 countries, they found that 69% of patients with depression reported only somatic symptoms. Hispanics, African Americans, and Asians were more likely to have somatic complaints in depression than Caucasians. The US Institute of Medicine’s 2002 report [8] and the US Surgeon General’s report [9] reiterate the need for each health system and clinician to have a process or system to examine cultural issues. Disparate access to care for racial and ethnic minorities is often based on socioeconomic status. In recent years, the annual number of suicide victims is over 30,000 in Japan [10]. In 2006, the suicide mortality rate was 24.2 per 100,000 for the whole population and 169.3 per 100,000 for persons aged 75 years old and older [10]. Although the Japanese population leads the world in longevity, it has the sixth highest rate of suicide. Most of these suicides are considered to be caused by depressive disorders. (Figs. 1, 2).

The S allele frequency of 21 Caucasian populations and three Asian populations among controls. The center of a box and the horizontal line (logarithm) indicate the prevalence and the 95% confidence interval (CI) in each study, with the areas of the boxes representing the weight of each study. The summary ORs of Caucasians and Asians based on the random effects model are 42.5% (95% CI = 40.8–44.2) and 76.8% (95% CI = 73.9–79.7), respectively. The summary OR is shown by the dotted vertical line. Statistical heterogeneity between studies among Caucasians and Asians were assessed as Q = 43.7, p = 0.002 and Q = 2.60, p = 0.27), respectively, by Cochran’s Q test

Meta-analysis of the 5-HTTLPR and major depressive disorder according to DSM IV among 17 Caucasian populations. The center of a box and the horizontal line (logarithm) indicate the odds ratio (OR) and the 95% confidence interval (CI) in each study, with the areas of the boxes representing the weight of each study. The summary OR based on the random effects model is represented by the middle of a diamond whose width indicates the 95% CI. The summary OR is shown by the dotted vertical line. Statistical heterogeneity between studies was assessed with Cochran’s Q test (Q = 12.1, p = 0.74). Summary OR = 1.31 (95% CI = 1.14–1.52). SS + LS versus LL
The prevalence of major depressive disorder is much lower than the heritability of bipolar disorder or schizophrenia. The heritability of major depressive disorder is likely to be in the range of 31–42%. This is probably at the lower end of the range and the level of heritability is likely to be substantially higher for reliably diagnosed major depressive disorder and subtypes such as recurrent major depressive disorder. In comparison, the heritabilities of schizophrenia and bipolar disorder are estimated to be approximately 70% [11]. Major depressive disorder is caused by multiple genes and does not follow Mendel’s laws of inheritance. Major depressive disorder, a common disease, results from a complex interplay of genes and environmental risk factors as well as other common multifactorial diseases such cancer and diabetes mellitus and cardiovascular disease. Several excellent reviews have been written on the topic of stress and depression in the past 15 years [12–18]. Environmental factors, such as pre-natal factors, loss, deprivation, grief, stress, natural disasters, war, social support systems, nutrition, exercise, drug effects, and medical illness, have been linked to major depressive disorder [19]. As for genetic factors, only minor susceptibility genes have been reliably identified (Table 1). The serotonin system provides a logical source of susceptibility genes for depression, because this system is the target of selective serotonin reuptake-inhibitor drugs that are effective in the treatment of depression. The 5-hydroxytryptamine (serotonin) transporter (5-HTT) has received particular attention, because it is involved in the reuptake of serotonin at brain synapses. One common polymorphic variant of the 5-HTT-linked polymorphic region (5-HTTLPR), which affects the promoter of the 5-HTT gene, causes reduced uptake of the neurotransmitter serotonin into the presynaptic cells in the brain [20]. However, despite strong biologic plausibility, the relationship between 5-HTTLPR and depression risk is unclear. Several studies have reported an association of the 5-HTTLPR polymorphism with depression risk, although other studies reported mixed or conflicting results. The individual results of these studies have been inconsistent and definite conclusions are difficult to establish. A reliable method for assessing individual studies and generating combined results is provided by systematic reviews using meta-analytical techniques. In this paper, we carried out a systematic review by systematically retrieving, assessing, and combining individual studies investigating the association between depression and a functional genetic polymorphism of 5-HTTLPR.
Polymorphic variants
With the cloning of the human genome, it has become apparent that genetic anomalies are not limited to high-risk groups. More than ten million common genetic variants exist and are referred to as polymorphisms. There are two major types of polymorphisms: tandem repeats and SNPs. Tandem repeats are an array of consecutive repeats. They include minisatellites (also known as variable number of tandem repeats), whose repeat units range from 9 to 80 bp, and microsatellites (also known as short tandem repeats), whose repeat units consist of only 1–6 bp. A SNP is a base pair within the genome that varies between two or more nucleotides. SNPs can alter gene function in a variety of ways: nonsynonymous variants alter function by changing the amino acid sequence of a gene, variants within the promoter alter transcription of a gene, and variants in close proximity to intron–exon boundaries alter splicing. The vast majority of polymorphisms have no functional significance; however, a few can influence tumor development or progression. Importantly, since many of these susceptibility polymorphisms are common in the population at large, they have the potential to influence the pathogenesis of depression in a large percentage of the population. Polymorphisms are present in germline DNA and are different from somatic mutations.
Prevalence of depression
Depressive disorders, which are syndromes characterized by the impairment of mood regulation, most commonly include major depressive disorder and dysthymia, a disorder characterized by chronic low mood [2, 21]. According to DSM-IV [2], the diagnosis of major depressive disorder can be made if a patient has five or more symptoms (depressed mood, loss of interest or pleasure in previously pleasurable activities, significant weight gain or loss, insomnia or hypersomnia, psychomotor agitation or retardation, fatigue, feelings of worthlessness or inappropriate guilt, impaired concentration and recurrent thoughts of death) during the same two-week interval with at least one of the symptoms being either depressed mood or loss of interest or pleasure in activities that were previously pleasurable. Not all patients will suffer from every symptom. The severity of symptoms will vary widely among individuals. Depression typically begins in the late 20s, but it can arise at any age, affecting everyone from young children to older adults. Major depressive disorder is common, costly, and disabling and is one of the most burdensome disorders worldwide [22–24]. Worldwide, it is the fourth leading cause of disability and the leading cause of nonfatal disease burden, accounting for almost 12% of total years lived with disability [25].
Mental disorders have been found to be common, with over a third of people in most countries reporting sufficient criteria at some point in their life [26]. The WHO estimated that about 450 million people worldwide currently suffer from some form of mental or behavioral disorder [27]. The WHO is currently undertaking a global survey of 26 countries in all regions of the world, based on International Statistical Classification of Diseases and Related Health Problems (ICD) and Diagnostic and DSM criteria [28]. The prevalence of metal disorders may be an underestimate, due to poor diagnosis (especially in countries without affordable access to mental health services) and low reporting rates, in part because of the predominant use of self-reported data, rather than semi-structured instruments such as the Structured Clinical Interview for DSM-IV (SCID).
In recent community surveys, 4–10% of the general population has experienced an episode of major depressive disorder within the past year [29–33]. There is strong evidence that the risk of depression is greater in women than in men [34–36]. A review of pooled surveys of mood disorders in different countries found 12-month prevalence rates of 4.1% (95% CI = 0.4–6.2) for major depressive disorder [4.9% (95% CI = 3.3–7.1) for men and 10.0% (95% CI = 6.4–14.6%) for women]. The average lifetime prevalence found was 6.7% (95% CI = 4.2–10.1) for major depressive disorder [3.8% (95% CI = 2.4–5.8) for men and 7.5% (95% CI = 4.5–11.3) for women] [37]. Similarly, women are about twice as likely as men to be diagnosed and treated for major depressive disorder in the United States [38]. Approximately 20–25% of women and 12% of men will experience a serious depression at least once in their lifetimes. Among children, depression appears to occur in equal numbers of girls and boys. However, as girls reach adolescence, they tend to experience greater rates of depression than boys [39]. This gender difference continues into older age. There are several theories for this gender disparity. Women may suffer from unique forms of depression related to their unique biology and life experiences and may be more likely than men to seek treatment. Hormonal factors may contribute to the increased rate of depression in women, particularly such factors as premenstrual changes, pregnancy, miscarriage, postpartum period, premenopause, and menopause. Many women face additional stresses, such as responsibilities at work and home, single parenthood, and caring for children and aging parents.
While women suffer from depression more often and attempt suicide more frequently, men are more successful in their suicide attempts [40]. There are several possible explanations for these contrasting patterns [41–43]. First, men use more lethal methods of suicide than women, so case fatality is higher. Second, women with depression are more likely than men to seek help from friends, family and health services. Last, it is possible that depression is more stigmatized in men than in women, so men may be less likely to report symptoms. Neverthless, evidence for this latter explanation is mixed [43, 44]. Furthermore, depressive symptoms are relatively common in older populations [45], especially among women [46].
Heterogeneity analysis demonstrated significant differences across 12-month and lifetime prevalence rates of depression among different ethnic populations. Large epidemiologic surveys [47, 48] find that compared with non-Hispanic whites, blacks have lower lifetime rates of depression and equivalent or lower rates of 12-month depression. At the same time, blacks are overrepresented in high-need populations (composed of individuals with multiple chronic illnesses that require regular medical monitoring, advising, or treatment), have reduced access to mental health services, and often receive poorer quality care than whites [49]. The US National Comorbidity Survey found that although blacks have a lower lifetime risk of mood disorder than whites, once diagnosed they were more likely to be persistently ill [50]. Ethnicity is a neglected dimension of the heterogeneity of the black population [51]. Although there are important commonalities in the black experience, there is also ethnic variation within the black population.
For studies conducted in Europe, the pooled 12-month prevalence rate was 8.0% (95% CI = 5.0–11.9) [37]. For studies conducted in Asia, pooled 12-month and lifetime rates were 0.85 (95% CI = 0.59–1.2) and 1.6 (approximately sevenfold lower than studies carried out in non-Asian countries [37]. The 12-month depression rates was highest among Hispanics (10.8%), followed by African Americans (8.9%) and Caucasians (7.8%) [52]. The World Mental Health (WMH) Survey Initiative aims to obtain accurate cross-national information about the prevalence and correlates of mental, substance, and behavioral disorders. Similarly, the WMH Japan Survey 2002–2003 showed that the 12-month prevalence rate was 2.9% (95% CI = 2.1–3.7) [53]. Another Japanese study by the Ministry of Education, Culture, Sports, Science and Technology reported that the 12-month and lifetime prevalence rates were 2.2 and 6.5%, respectively [54]. Elevated depression rates among minority individuals were largely associated with greater health burdens and lack of health insurance, both factors amenable to public policy intervention. Some studies report higher rates of major depressive disorder in African Americans compared with Caucasians, whereas others report lower or equivalent rates in African Americans [55]. The relationship between ethnicity and depression is complex.
The 12-month and lifetime prevalence rates based on DSM-IV criteria have been reported to be 2.2 and 6.5%, respectively, while the corresponding values based on ICD-10 criteria have been reported to be 2.2 and 7.5%, respectively [54]. Although studies using DSM-III-R criteria had pooled 12-month and lifetime prevalence rates that were around 3–4 times higher than those for studies using other diagnostic criteria [37], there was not a large difference between ICD-10 and DMS-IV criteria.
Stress, a risk factor for depression
Stress is a term that means different things to different people, but generally has a negative connotation [56]. Yet, stress is a familiar aspect of modern life and is a stimulant for some individuals, but a problem for many others. Stress has been defined as a constellation of events, which begins with a stimulus (stressor) that precipitates a reaction in the brain (stress perception), which subsequently activates physiologic systems in the body (stress response) [57, 58]. The physiologic stress response results in the release of neurotransmitters and hormones that serve as the brain’s messengers to the rest of the body. The consequences of this physiologic response are generally adaptive in the short run [57, 59], but can be damaging when stress is chronic and long lasting [57, 60].
Chronic stress is long term, negative stress that has become detrimental to the individual, while acute stress is short term, positive stress, which is relatively harmless to the individual and often gives an extra burst of energy that is needed during the stressful period. An important marker of the deleterious effects of chronic stress is a breakdown in the regulation of the circadian corticosterone rhythm in rodents and cortisol rhythm in humans [57, 61]. Chronic stress seems to be linked to the etiology of many diseases. A number of studies have shown that stress can be immunosuppressive, and hence that it can be detrimental to health. Moreover, glucocorticoid stress hormones are regarded widely as immunosuppressive [62] and are used clinically as anti-inflammatory agents [63]. Generally, it has been accepted that stress and stress mediators can have potent immunosuppressive and health adverse effects.
Continuing adverse conditions, such as poverty, medical disabilities, and lasting marital discord, have been found to be associated with an increased risk of depression [64–67]. Breslau and Davis [68] compared chronic stress in women to having a disabled child, and found such experiences to be linked to more lifetime episodes of major depressive disorder but not to higher rates of current depression in such women compared with non-stressed controls. Chronic stress, defined as an absence of social support, also was found to be associated with depression [69]. Chronic stresses, such as illness, lack of social support, divorce, loss of a loved one and loss of employment, have been considerably associated with an increased risk of depression, but its definition has varied.
Chronic stress is an important area for further study, with significant implications for interpretation of associations between episodic stress and depression. Unresolved empirical questions remain, as do conceptual questions of how chronic and episodic stressors may work together to produce depression and considerably more effort is needed to refine the definitions and methods of assessment of chronic stress.
Genetic epidemiology
Genetic influences have been supported by adoption studies and twin studies of major depressive disorder. The heritability of major depressive disorder is likely to be in the range of 31–42% [11]. Similarly, heritability based on twin studies is 40–50% [70–75] and adoption studies also support a role for genetic factors [76–78]. In comparison, the heritabilities of schizophrenia and bipolar disorder are estimated to be approximately 70% [11]. The OR (first-degree relatives of depression probands vs. the general population) has been reported to be around 2–3 [79–81]. Genetic factors have been implicated in the etiology of depression and many studies have determined that changes in protein structure correlate with a predisposition to specific conditions. More than ten million SNPs have been identified in humans; however, the importance of most SNPs for health and disease is not understood. Most SNPs are indeed unimportant and because of often inadequately powered studies; many observations on SNP effects cannot be repeated by other researchers. SNPs are at best shown to influence protein function or level, rarely to influence risk of disease, and almost never to influence total mortality, the ultimate endpoint. However, specific genes or relevant DNA sequence variations involved in the pathogenesis of depression have not yet been identified.
The monoamine hypothesis proposes that depression is due to a deficiency in monoaminergic neurotransmission. Developed out of a meticulous research effort in modern psychiatry, the monoamine hypothesis is an early milestone in the field of depression. Under this hypothesis, depression is postulated to reflect a deficiency or imbalance in noradrenaline [82, 83] or serotonin [84]. These conclusions were based on observations that several antidepressant drugs increased synaptic concentrations of noradrenaline or serotonin, and that reserpine, a catecholamine depleting drug, could cause depression-like symptoms [82, 83]. Catecholamine depletion by dietary methods has also been shown to induce a relapse of depressive symptoms [84–87]. Subsequent variations of the monoamine hypothesis were supported by animal research, including the findings that antidepressants effectively treat learned helplessness. Currently, the evolving monoamine hypothesis considers the possibility that depression may be linked to a deficiency in signal transduction from the monoamine neurotransmitter to its postsynaptic neuron in the presence of normal amounts of neurotransmitter and receptor. Such a deficiency in the molecular events that cascade from receptor occupancy by the neurotransmitter to transcription of genes could lead to a deficient response of target neurons to neurotransmission and thus, depression. Selected candidate genes are shown in Table 1.
The 5-HTT gene regulates brain serotonin neurotransmission by removing the neurotransmitter from the extracellular space. Since the development of the selective serotonin reuptake-inhibitors, a putative role for 5-HTT in the etiology of depression has been explored. The discovery of a functional 5-HTT polymorphism has provided a novel tool to further scrutinize the role of serotonergic neurons in depression. A repeat of 20–23 base pairs has been observed as a motif within a polymorphic region of the 5-HTT gene, and it occurs as two prevalent alleles: one consisting of 14 repeats (S allele) and another of 16 repeats (L allele). This functional polymorphism in the promoter region, termed 5-HTTLPR, alters transcription of the serotonin transporter gene. The S allele leads to less transcriptional efficiency of serotonin [88, 89], and it can partly account for anxiety-related personality traits [20].
Another common polymorphism is a variable number tandem repeat (VNTR) in intron 2 (STin2), which has three alleles consisting of either 9 (STin2.9), 10 (STin2.10) or 12 (STin2.12) repeats, were shown to be in linkage disequilibrium, with the positive association between the STin2 allele 10 and the 5-HTTLPR L allele [90]. Variation at the VNTR can also influence expression of the transporter with the polymorphic VNTR regions acting as transcriptional regulators [91] although it is likely to have no significant effect on function.
Tryptophan hydroxylase (TPH) is the enzyme involved in the biosynthesis of serotonin. It catalyzes the oxygenation of tryptophan to 5-hydroxytryptophan, which is decarboxylated to serotonin [92]. It is thus the rate-limiting enzyme in the biosynthesis of serotonin. Changes in the metabolism of the essential amino acid tryptophan play an important role in the brain–endocrine-immune system interaction that is hypothesized to be involved in the pathophysiology of major depressive disorder. There are two main pathways of tryptophan metabolism: one is the serotonin pathway and the other is the kynurenine pathway [93]. Dysfunction of the serotonergic system has been hypothesized to play an important role in depression. The higher serotonergic activity during stress leads to a higher breakdown of serotonin. Sustained stress will lead to diminution of serotonin (synthesis < degradation). Thus, chronic stress may lead to functional shortage of the supply of serotonin.
Two genes have been discovered which encode for the TPH isoforms (TPH1 and TPH2). Most of the molecular genetic studies on the TPH genes so far have examined the two polymorphisms of the TPH1, A218C and A779C, which have been found to be in complete or strong linkage disequilibrium in previous studies [94]. A polymorphism in intron 7 of TPH1, A218C has been extensively examined in association studies, and the A allele is associated with anxiety symptoms in depression [95]. There is no association between the TPH2 polymorphisms and stress induced depression [96, 97].
Catechol-O-methyltransferase (COMT) is an important enzyme involved in the degradation of catecholamine neurotransmitters. Depression is thought to involve, in part, dysregulation of serotonergic neurotransmission. A Val158Met polymorphism affects the activity of the COMT enzyme and individuals with the Val/Val genotype have a 3 to 4-times higher enzyme activity than those with the Met/Met genotype [98]. A large European study reported an association between the Val/Val genotype and early onset major depressive disorder [99], but conflicting results have been found in other studies with fewer participants [100].
As serotonin exerts its effect on specific receptors on the postsynaptic membranes, the serotonin receptor (5-HTR) genes are considered good candidates. The 5-HTRs affect the release and activity of other neurotransmitters such as glutamate, dopamine and gamma-aminobutyric acid (GABA). There are seven main types of 5-HTRs. Including subtypes, there are a total of at least 14 different receptors based on their pharmacological responses to specific ligands, sequence similarities at the gene and amino acid levels, gene organization, and second messenger coupling pathways [101]. Several subtypes, including the 5-HTR1A, 5-HTR1B, 5-HTR2A, 5-HTR3 and 5-HTR4, act to facilitate neurotransmitter dopamine release, while the 5-HTR2C receptor mediates an inhibitory effect of serotonin on dopamine release. Most 5-HTR subtypes only modulate dopamine release when serotonin and/or dopamine neurons are stimulated, but the 5-HTR2C, characterized by high levels of constitutive activity, inhibits tonic as well as evoked dopamine release [102]. The 5-HTR1A is an important member of the large family of serotonin receptors [103]. The 5-HTR1A is located both at a post-synaptic and at a pre-synaptic level; in the first case, they mediate the action of serotonin on cortical and limbic neurons, and in the second case, they act as serotonergic auto-receptors on serotonergic neurons in the raphe nuclei and prevent the release of serotonin by negative feedback [104, 105]. In depressed individuals, the number of serotonin receptors, including the 5-HTR1A autoreceptors, is increased [106]. A common functional polymorphism C-1019G in the promoter region of the human 5-HT1A receptor gene has been reported, which may be useful in identifying psychopathology associated with altered function of the human 5-HT1A receptor [107].
Mounting evidence shows that brain derived neurotrophic factor (BDNF) plays a crucial role in synaptic plasticity. Normally, BDNF sustains the viability of brain neurons. Under stress, however, the gene for BDNF is repressed, leading to atrophy and possible apoptosis of vulnerable neurons in the hippocampus when their neurotrophic factor BDNF is absent. These events in turn lead to depression and to the consequences of repeated depressive episodes, namely, more and more episodes and less and less responsiveness to treatment. Due to its potential involvement in psychiatric diseases like depression, BDNF became a major target in research. A functional polymorphism of the BNDF Val66Met influences and reduces trafficking and secretion of BNDF protein in the brain and is thought to be associated with low BDNF levels in major depressive disorder [108–110]. Some did associate the BDNF Val66Met genotype with depression [111], but other groups found only weak or no associations [112–115].
The norepinephrine transporter (NET), a Na+/Cl-dependent substrate specific transporter, terminates noradrenergic signalling by rapid re-uptake of neuronally released norepinephrine into presynaptic terminals. NET exerts a fine regulated control over norepinephrine-mediated physiological effects such as depression. As the 5′ flanking promoter region of the NET gene, NET T-182C, contains several cis elements that play a critical role in transcription regulation [116, 117], changes in this promoter DNA structure may lead to an altered transcriptional activity responsible for a predisposition to major depressive disorder [118]. Although a silent G1287A polymorphism, located at exon 9 of the NET gene, is not an important factor in susceptibility to depression in a Japanese population [119], it cannot be denied that it may be an important candidate.
DOPA decarboxylase is an enzyme implicated in two metabolic pathways, synthesizing two important neurotransmitters, dopamine and serotonin [120]. Following the hydroxylation of tyrosine to form 3,4-dihydroxy-l-phenylalanine (l-DOPA), catalyzed by tyrosine hydroxylase (TH), DDC decarboxylates l-DOPA to form dopamine. The TH is the initial and rate-limiting enzyme in the biosynthesis of catecholamine neurotransmitters and has been a candidate for possible involvement in the development of psychiatric illness [121]. Among polymorphisms in the TH gene identified so far, a penta-allelic polymorphism consisting of several numbers of tetranucleotide repeats [or microsatellite polymorphism (TCAT) n ] in intron 1 and three SNPs, namely Val81Met, Leu205Pro and Val468Met, may alter the functional activity of TH protein. A tetranucleotide repeat in intron 1 has been reported to play a role in psychiatric illnesses [122–125].
Dopamine is the most abundant catecholaminergic neurotransmitter in the brain and is involved in the regulation of emotions, motivation, reward, and reinforcement behavior, through the mesocorticolimbic pathway [126]. Preclinical and clinical studies incriminate the dopaminergic system in affective disorders. While hyperfunction of the dopaminergic system may lead to manic behavior, its hypofunction may lead to depressive symptoms [127, 128]. Dopamine, functioning at dopamine D1-like (D1 and D5) and D2-like (D2, D3 and D4) receptors, crucially influences both the induction and the reversal of neuroplasticity at corticostriatal synapses.
These two subfamilies have different pharmacologic properties, signal transduction properties, and genomic organization. The receptors of the D1-like subfamily stimulate cAMP synthesis through coupling with Gs-like proteins, and their genes do not contain introns within their protein coding regions. The receptors of the D2-like family inhibit cAMP synthesis through their interaction with Gi-like proteins and share a similar genomic organization, which includes introns within their protein coding regions [129]. On the basis of sequence similarity, the G subunits have been divided into four families (Gi, Gs, Gq and G12). Heterotrimeric guanine nucleotide-binding proteins (G proteins) are signal transducers and are attached to the cell surface plasma membrane that connects receptors to effectors and thus to intracellular signaling pathways [130]. Receptors that couple to G proteins communicate signals from a large number of hormones, neurotransmitters, chemokines, and autocrine and paracrine factors. Many important hormones and neurotransmitters, including epinephrine, acetylcholine, dopamine and serotonin, use the Gi and Go pathway to evoke physiological responses.
In vitro and in vivo studies suggested that the −141C Ins/Del polymorphism may be directly responsible for the regulation of DRD2 expression and DRD2 function [131, 132]. Ser311Cys in DRD2 has been reported to be associated with a reduced ability of activating the appropriate Gi-like protein [133]. Brain imaging studies of healthy volunteers have shown that individuals with an A1 allele (C allele of C32806T) of DRD2 have a reduced number of dopamine D2 receptors [134, 135]. The DRD3 Ser9Gly genotypes were reported to differ with regard to their dopamine-binding affinity [136]. The most widely studied polymorphism of DRD4 is a 48 bp variable number of tandem repeats (VNTR) which encodes for the third intracytoplasmic loop and contains 2–11 repeats [137]. A 48 base pairs repeat in exon 3 of DRD4 was associated with the potency of inhibition of cAMP formation by dopamine [138]. The DRD4 48 base pairs repeat in the exon 3 was significantly associated with major depression while the DRD2 Ser311Cys and DRD3 Ser9Gly polymorphisms were not [139]. These polymorphisms may be hypothesized to influence major depressive disorder. These polymorphisms have been reported to increase the susceptibility to depression, although negative findings have also been reported.
Major depressive disorder and 5-HTTLPR polymorphism
The first study suggests that variation at 5-HTTLPR may influence susceptibility to major depressive disorder [90]. Three case-control studies were separately carried out in the British, German and Italian populations. A significant association between the presence of the S allele and major depressive disorder, whether by allele (population-adjusted OR = 1.23, 95% CI = 1.02–1.49; the S allele vs. the L allele) or genotype (population-adjusted OR = 1.53, 95% CI = 1.04–2.23; the SS genotype vs. the LL genotype), was observed in a combined European sample of three populations although each study found no significant association [90].
The S allele is a disease allele; thus, the authors calculated ORs for the presence of one (S allele carrier) or two (S/S genotype) S alleles compared with the LL genotype. However, the second study found that the SS genotype or S allele was nonsignificantly associated with decreased risk of depression in the British population [140]. A nonsignificant protective effect of the S allele was observed among Spaniards [109] and Germans [141, 142]. Five studies of whites [143–147] found no substantial relationships between the 5-HTTLPR genotype and major depressive disorder. In contrast, seven Caucasian [148–154] and one non-Ashkenazi Jewish [144] studies found that the SS genotype was associated with a modest increase in risk of depression. Furthermore, one Polish [155] and one German [156] studies found that the SS genotype was associated with a significant increase in risk of depression. Japanese studies did not find any significant difference between patients with major depressive disorder and controls [157, 158]. Likewise, the frequency of the S allele did not differ between the normal subjects and the depressed patients in a Korean population [159].
Two meta-analyses on the association between 5-HTTLPR and major depressive disorder have been published in 2004 [160] and 2005 [161], respectively. In the first meta-analysis based on ten case-control studies, the number of depressed cases and controls was 910 and 2,017, respectively. The first meta-analysis showed that individuals with the SS genotype had a 16% (95% CI = 1.03–1.31) increased risk of major depressive disorder compared with individuals with the LL genotype. The results supported the hypothesis that individuals with the SS genotype are at higher risk of developing major depressive disorder.
Five individual studies [90, 139, 148, 149, 157] were included in both meta-analyses. The second meta-analysis was somewhat different from the first one, because ORs for the S allele compared with the L allele were summarized. The second meta-analysis based on ten case-control studies, comprised of 1,961 cases and 3,402 controls, showed that the summary OR was close to unity [OR = 1.05, 95% CI = 0.96–1.14].
We conducted MEDLINE, Current Contents and Web of Science searches using “depression”, “5-HTTLPR” and “polymorphism” for papers published before March 2008. Additional articles were identified through the references cited in the first series of articles selected. Articles included in the meta-analysis were in any language, involved human subjects, published in the primary literature and had no obvious overlap of subjects with other studies. We excluded studies with the same data or overlapping data by the same authors. Case-control studies were eligible if they had determined the distribution of the relevant genotypes in depression cases and in concurrent controls using a molecular method for genotyping. Using the MEDLINE database, we identified 22 genetic epidemiological studies that provided information on depression associated with the 5-HTTLPR polymorphism. No additional articles through Current Contents or Web of Science were identified. Data were combined using random effects (DerSimonian and Laird method) model [162].
In our unpublished meta-analysis, the 22 case-control studies in 25 different ethnic populations of major depressive disorder and 5-HTTLPR included 7,919 subjects (2,934 depressed cases and 4,985 controls). Our meta-analysis (unpublished) did not include a large European collaborative study of Mendelwics et al. [163] due to the absence of information on genotype frequency. The summary OR for the SS genotype was 1.34 (95% CI = 1.14–1.57). Our results (unpublished) were robust in sensitivity analyses that were restricted to studies of Caucasians (OR = 1.34, 95% CI = 1.12–1.61) or studies of Caucasians based on DSM-IV criteria (OR = 1.34, 95% CI = 1.08–1.66). The summary OR for the SS genotype compared with the combined genotype among Caucasian studies based on DSM-IV was 1.31 (95% CI = 1.14–1.52). Thus, the S allele would act in a recessive fashion. Statistically significant heterogeneity was not seen in this sensitivity analysis. Similarly, the summary OR for the combined genotype among Asian studies based on DSM-IV was 1.40 (95% CI = 0.84–2.31).
Summary frequencies of the S allele among Caucasians and Asians based on random effects model were 42.5% (95% CI = 40.8–44.2%) and 76.8% (95% CI = 73.9–79.7%), respectively (Table 2). The distribution of the S allele was significantly different between Asians and Caucasians (p < 0.001).
Discussion
When Caucasian populations were restricted to the studies based on DSM-IV, the summary OR for the SS genotype was 1.34 (95% CI = 1.08–1.66). Thus, the 5-HTT gene appears to be a candidate for major depressive disorder susceptibility genes in Caucasian populations. When Asian populations (all Japanese) were restricted to the studies based on DSM-IV criteria, the summary OR for the SS and LS genotypes combined were 1.40 (95% CI = 0.84–2.43). Our meta-analysis (unpublished) did not support a major role of 5-HTTLPR in major depressive disorder among Asians although there are only a small number of available articles regarding Asian populations. The ethnic difference of the association between this polymorphism and depression is not clear. Generally, low frequency of the “at risk” genotype reduces the statistical power. As the distribution of the S allele was significantly different between Asians and Caucasians (p < 0.001), this is not the case. Given the higher frequency of the S allele of the 5-HTTLPR in Japanese subjects, if this allele is associated with an increased risk of depression then the prevalence of major depressive disorder would be higher among Japanese than Caucasians. Major depressive disorder is a multifactorial disease that results from complex interactions between many genetic and environmental factors. Thus, if the S allele of the 5-HTTLPR increases the susceptibility to major depressive disorder, ethnic differences in roles of the polymorphism may be caused by gene–gene interactions, different linkages to the polymorphisms determining depression risk and different lifestyles.
Identification of gene-environment and gene–gene interactions has become increasingly important in understanding psychiatric disorders. According to a model of gene–gene interaction, it has been shown that subjects with an increasing number of ‘at-risk’ variant alleles for DNA repair genes have increasing levels of DNA damage, with a dose–response relationship [164]. These observations from a large pooled analysis strongly suggest the existence of gene–gene interactions in lung carcinogenesis. Although it is hypothesized that gene-gene interactions among genetic variants increase the individual risk of depression, the role of gene–gene interaction in the risk of depression is not clear. The study of gene–gene interactions may be important in the identification of high-susceptibility subgroups. No studies have investigated gene–gene interactions in depression. As a rule of thumb, the study of the interaction between two genes needs a study size four times higher than for the study of the main effect of a single gene. Advances in identification of new variants and in high-throughput genotyping techniques will facilitate analysis of multiple polymorphisms within the genes along the same pathway [165]. Therefore, it is likely that definitive studies in the future will require analysis of large samples of cases and controls [166, 167].
In addition to adequate sample size, assessment of gene–gene interaction also depends upon the proper statistical evaluation of interaction on the multiplicative and additive models. Well-organized studies with a large sample size are needed to help further illuminate the complex landscape of major depressive disorder risk and genetic variations.
Ogilvie et al. [168] first demonstrated that there was an excess of the 9-repeat allele of another 5-HTT STin2 polymorphism in major depressive disorder patients in comparison to controls. However, this finding was not replicated in other Caucasian [139, 143, 148, 149, 169–172] or an Asian study [157] using independent samples. The rarity of the STin2.9 allele (below 1%) means that the risk attributable to this allele is small. Liu et al. [173] demonstrated that the 12-repeat allele was associated with major depressive disorder in a Chinese population. Although the 5-HTTLPR may play a pathogenic role, it seems more likely that it is in linkage disequilibrium with polymorphisms other than the STin2 polymorphisms within or close to the 5-HTT gene. Recently developed haplotype-based methods can be anticipated that will facilitate the evaluation of haplotypic effects, including selected physically close polymorphisms.
Although the risk associated with the 5-HTTLPR polymorphism may be small compared to that conferred by high-penetrance disease genes, their public health implication may be large, because of their high frequency in the general population. It is essential that epidemiological investigations of monoaminergic polymorphisms are adequately designed. Unfortunately, a fairly large number of studies are limited by their sample size and, consequently, low power to detect effects that may truly exist. Also given the borderline significance of some associations and multiple comparisons, there is a possibility that one or more of these findings are false-positives [174]. Large and combined analyses such as those by Healey et al. [175] and Spurdle et al. [176] are preferred to minimize the likelihood of both false-positive and false-negative results. In addition, the choice of the control group is a key issue in case-control studies. Controls should be a representative sample of the source population which generated the cases; in practice, the choice of controls has been quite variable. Hospital controls are usually included in genetic association studies because of their higher response rate and compliance to the protocol and the easier access to biological samples [177]. Although the case-control genetic association study differed in the choice of the control group, no differences in allele frequencies were seen by age or type of controls [178]. The general lack of a significant association between allele frequency and age may allow epidemiologists to rule out the possibility that polymorphisms are determinants of longevity. The lack of any effect of choice of controls is important for combining data from different case-control studies that use one or the other source for the control population. Furthermore, only studies with allelic frequencies being in the Hardy–Weinberg equilibrium (Pearson χ2 test, p ≥ 0.05) were combined in meta-analyses because departure from HWE can imply possible ethnic admixture in the population or selection bias (lack of representativeness of the general population). The choice of the control group may be less crucial in the case-control genetic association studies. Controls should be chosen in such a way that, if they were cases, they would be included in the case group; when controls are matched to cases, it is essential to account for matching in the analysis. When appropriate, confounding should be controlled for with particular consideration to race and ethnic group. An additional major concern is the grouping of genotypes for calculation of ORs; without functional data to dictate genotype groupings, it seems prudent to present two ORs per polymorphism (one for heterozygotes vs. common-allele homozygotes and one for rare-allele homozygotes vs. common-allele homozygotes) so that dominant, codominant, or recessive patterns may be elucidated.
Evidence for an association between the 5-HTTLPR genotypes and depression is inconclusive. Caspi et al. [179] reported that there were no significant differences between the 5-HTTLPR genotypes in the number of life events they experienced. They reported that stressful life events predicted a diagnosis of major depressive disorder among carriers of the S allele but not among those with the LL genotype. Thus the study provides evidence of a gene–environment interaction, in which an individual’s response to environmental insults is moderated by genetic susceptibility. Several studies report that depressive symptoms are increased in 5-HTTLPR S-allele carriers who report a history of adverse or stressful life events [180–186]. Wilhelm et al. [186] reported that adverse events appeared to protect against depression in the subjects with LL genotype. Interestingly, a similar trend is observed in other studies [179, 180, 184] that have reported an interaction, although an opposite effect of adversity on depression risk across genotypes seems biologically unlikely. The study of Kaufman et al. [181] specifically reported increased rates of depression in adult carriers of the S allele who had been abused in childhood. However, Gillespie et al. [187] and Surtees et al. [188] found no evidence to support a main effect of 5-HTTLPR, or an interaction between the 5-HTTLPR genotype and stressful life events on major depressive disorder. Evidence for the role of the gene–environment interaction with childhood events is much stronger than that for the interaction with life events occurring close to the onset of depression [189]. Later adult stressful events may have served as a marker for childhood events. Prospective studies may fail to replicate gene-environment interaction if respondents already suffer from depression at baseline. Most studies found evidence for a gene–environment interaction although the studies have examined different stress measures such as stressful life events (illness, death of a relative, marital breakdown, financial crisis, unemployment) [179, 180, 183, 184, 186, 187], parental educational level [180], social adversity (problems relating housing, work, relationships and social difficulties) [180, 188], childhood maltreatment (physical abuse, sexual abuse, neglect, emotional abuse, domestic violence) [179, 181] and family constellation, type of residence, family relationship and psychosocial index [185] and different assessment methods (scales) of stress measures such as a life-history calendar [179], the Social Problems Questionnaire [180], the List of Threatening Experience [180, 187], the Arizona Social Support Interview Schedule [181], long-term contextual threat scale [182], the St Paul–Ramsey Scale [184] and the Health and Life Experiences Questionnaire [186, 188]. The severity of stress measures was assessed by the number of the life events [179, 181, 183, 186–188] or dichotomized groups [high/low (dichotomized at the median), no/one or more, or no and one/two or more] [180, 182, 184, 185]. Additionally, there is some evidence that women may be more involved in gene–environment interaction [180, 183, 185]. Although this evidence is only suggestive, it should be focused on in studies of depression in women. Again, the interaction between the 5-HTTLPR genotype and the life events on major depressive disorder may exist although there were marked differences of factors associated with stress measures among the studies. If the gene–environment interaction can be confirmed by further adequately powered and well-designed studies, it will provide an intriguing base for further exploration and dissection of pathological mechanisms underlying depression.
Continued advances in SNP maps and in high-throughput genotyping methods will facilitate the analysis of multiple polymorphisms within genes and analysis of multiple genes within pathways. The effects of polymorphisms are best represented by their haplotypes. Data from multiple polymorphisms within a gene can be combined to create haplotypes, the set of multiple alleles on a single chromosome. None of the studies reviewed here reported haplotype associations, although several studies analyzed multiple polymorphisms within a gene, sometimes with inconsistent results. The analysis of haplotypes can increase power to detect disease associations because of higher heterozygosity and tighter linkage disequilibrium with disease-causing mutations [190–192]. In addition, analysis of haplotypes offers the advantage of not assuming that any of the genotyped polymorphisms is functional; rather, it allows for the possibility of an ungenotyped functional variant to be in linkage disequilibrium with the genotyped polymorphisms [193].
An analysis of data from multiple genes within the same DNA-repair pathway (particularly those known to form complexes) can provide more comprehensive insight into the studied associations. Such an analysis may shed light on the complexities of the many pathways involved with monoaminergic pathway and depression development and provide hypotheses for future functional studies. Because of concerns over inflated type I error rates in pathway-wide or genome-wide association studies, methods of statistical analysis seeking to obviate this problem are under development [194]. The ability to include haplotype information and data from multiple genes, and to model their interactions, will provide more powerful and comprehensive assessments of the monoaminergic pathways.
Although the summary risk for developing major depressive disorder in individuals of each genotype may not be large, major depressive disorder is such a common disease that even a small increase in risk can translate to a large number of excess major depressive disorder cases. Therefore, polymorphisms, even those not strongly associated with major depressive disorder, should be considered as a potentially important public health issue. In addition, a susceptibility factor in one population may not be a factor in another. There are differences in the prevalence of the 5-HTTLPR polymorphisms across populations. In a population where the prevalence of an “at-risk” genotype in a given polymorphism is very low, the “at-risk” allele or “at-risk” genotype may be too infrequent to assess its associated risk. At a population level, the attributable risk must be small simply because it is an infrequent allele. Finally, the major burden of major depressive disorder in the population probably results from complex interaction between many genetic and environmental factors over time. Further investigations of the combined effects of polymorphisms between monoaminergic genes and genes listed in Table 1 may help to clarify the influence of genetic variation in the process of developing the depressive state. Consortia and international collaborative studies, which may maximize study efficacy and overcome the limitations of individual studies, are needed to help further illuminate the complex landscape of major depressive disorder risk and genetic variations.
References
http://www.who.int/mental_health/prevention/suicide/suicideprevent/en/.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4th ed. Washington: American Psychiatric Association; 1994.
Guze SB, Robins E. Suicide and primary affective disorders. Br J Psychiatry. 1970;117:437–8.
Kapur S, Mieczkowski T, Mann JJ. Antidepressant medications and the relative risk of suicide attempt and suicide. JAMA. 1992;268:3441–5.
Nierenberg AA, Gray SM, Grandin LD. Mood disorders and suicide. J Clin Psychiatry. 2001;62(Suppl 25):27–30.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM-IV-TR). 4th ed. Washington: American Psychiatric Publishing Inc; 2000.
Simon GE, VonKorff M, Piccinelli M, Fullerton C, Ormel J. An international study of the relation between somatic symptoms and depression. N Engl J Med. 1999;341:1329–35.
Institute of Medicine. Unequal treatment: confronting racial and ethnic disparities in health care (March 2002). http://www.iom.edu/report.asp?id=4475. Accessed 20 March 2002.
US Public Health Service. Substance abuse and mental health administration. Mental health: culture, race, and ethnicity; a supplement to mental health: a report of the surgeon general. http://www.surgeongeneral.gov/library/mentalhealth/cre/.
Japanese Ministry of Health, Labor and Welfare. Vital statistics of Japan 2005. Tokyo: Health and Welfare Statistics Association; 2006.
Kendler KS. Overview: a current perspective on twin studies of schizophrenia. Am J Psychiatry. 1983;140:1413–25.
Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214.
Mazure CM. Life stressors as risk factors in depression. Clin Psychol Sci Pract. 1998;5:291–313.
Monroe SM, Hadjiyannakis K. The social environment and depression: focusing on severe life stress. In: Gotlib IH, Hammen CL, editors. Handbook of Depression. New York: Guilford Press; 2002. p. 314–40.
Tennant C. Life events, stress and depression: a review of the findings. Aust NZ J Psychiatry. 2002;36:173–82.
Paykel ES. Life events and affective disorders. Acta Psychiatr Scand. 2003;108:61–6.
Hammen C. Stress and depression. Annu Rev Clin Psychol. 2005;1:293–319.
Belmaker RH, Agam G. Major depressive disorder. N Engl J Med. 2008;358:55–68.
Wong ML, Licinio J. Research and treatment approaches to depression. Nat Rev Neurosci. 2001;2:343–51.
Lesch KP, Bengel D, Heils A, Sabol SZ, Greenberg BD, Petri S, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–31.
Edgerton JE, Campbell RE, editors. American psychiatric glossary. 7th ed. Washington: American Psychiatric Press Inc; 1994.
Grant BF, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Arch Gen Psychiatry. 2004;61:807–16.
Goetzel RZ, Hawkins K, Ozminski RJ. The health and productivity cost burden of the “top 10” physical and mental conditions affecting six large US employers in 1999. J Occup Environ Med. 2003;45:5–14.
Murray CJ, Lopez AD. The global burden of disease: a comprehensive assessment of mortality and disability from diseases, injuries, and risk factors in 1990 and projected to 2020. Cambridge: Harvard University Press; 1996.
Ustun TB, Ayuso-Mateos JL, Chatterji S, Mathers C, Murray CJ. Global burden of depressive disorders in the year 2000. Br J Psychiatry. 2004;184:386–92.
WHO International Consortium in Psychiatric Epidemiology. Cross-national comparisons of the prevalences and correlates of mental disorders Bulletin of the World Health Organization v.78 n.4. 2000.
WHO, The world health report. http://www.who.int/whr/2001/chapter1/en/index.html.
Andrews G, Henderson S, Hall W. Prevalence, comorbidity, disability and service utilisation. Overview of the Australian National Mental Health Survey. Br J Psychiatry. 2001;178:145–53.
Bijl RV, Ravelli A, van Zessen G. Prevalence of psychiatric disorder in the general population: results of The Netherlands Mental Health Survey and Incidence Study (NEMESIS). Soc Psychiatry Psychiatr Epidemiol. 1998;33:587–95.
Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51:8–19.
Offord DR, Boyle MH, Campbell D, Goering P, Lin E, Wong M, et al. One-year prevalence of psychiatric disorder in Ontarians 15–64 years of age. Can J Psychiatry. 1996;41:559–63.
Regier DA, Boyd JH, Burke JD Jr, Rae DS, Myers JK, Kramer M, et al. One-month prevalence of mental disorders in the United States. Based on five epidemiologic catchment area sites. Arch Gen Psychiatry. 1988;45:977–86.
Evans K, McGrath J, Milns R. Searching for schizophrenia in ancient Greek and Roman literature: a systematic review. Acta Psychiatr Scand. 2003;107:323–30.
Weissman MM, Bland R, Joyce PR, Newman S, Wells JE, Wittchen H-U. Sex differences in rates of depression: cross-national perspectives. J Affect Disord. 1993;29:77–84.
Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey. I: lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29:85–96.
Gater R, Tansella M, Korten A, Tiemens BG, Mavreas VG, Olatawura MO. Sex differences in the prevalence and detection of depressive and anxiety disorders in general health care settings. Arch Gen Psychiatry. 1998;55:405–13.
Bennett DS, Ambrosini PJ, Kudes D, Metz C, Rabinovich H. Gender differences in adolescent depression: do symptoms differ for boys and girls? J Affect Disord. 2005;89:35–44.
Bernal M, Haro JM, Bernert S, Brugha T, de Graaf R, Bruffaerts R, et al. Risk factors for suicidality in Europe: results from the ESEMED study. J Affect Disord. 2007;101:27–34.
Hawton K. Sex and suicide. Gender differences in suicidal behaviour. Br J Psychiatry. 2000;177:484–5.
Biddle L, Gunnell D, Sharp D, Donovan JL. Factors influencing help seeking in mentally distressed young adults: a cross-sectional survey. Br J Gen Pract. 2004;54:248–53.
Bogner HR, Gallo JJ. Are higher rates of depression in women accounted for by differential symptom reporting? Soc Psychiatry Psychiatr Epidemiol. 2004;39:126–32.
Gunnell D, Rasul F, Stansfeld SA, Hart CL, Davey SG. Gender differences in self-reported minor mental disorder and its association with suicide. A 20-year follow-up of the Renfrew and Paisley cohort. Soc Psychiatry Psychiatr Epidemiol. 2002;37:457–9.
Haynie DA, Berg S, Johansson B, Gatz M, Zarit SH. Symptoms of depression in the oldest old: a longitudinal study. J Gerontol B Psychol Sci Soc Sci. 2001;56:P111–8.
Kasen S, Cohen P, Chen H, Castille D. Depression in adult women: age changes and cohort effects. Am J Public Health. 2003;93:2061–6.
Weissman MM, Bland RC, Canino GJ, Faravelli C, Greenwald S, Hwu HG, et al. Cross-national epidemiology of major depression and bipolar disorder. JAMA. 1996;276:293–9.
Blazer DG, Kessler RC, McGonagle KA, Swartz MS. The prevalence and distribution of major depression in a national community sample: the National Comorbidity Survey. Am J Psychiatry. 1994;151:979–86.
Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. National Comorbidity Survey Replication. The epidemiology of major depressive disorder. JAMA. 2003;289:3095–105.
US Department of Health and Human Services. Mental health: culture, race, and ethnicity: a supplement to mental health: a report of the surgeon general—executive summary. Rockville: US Dept of Health and Human Services, Public Health Service, Office of the Surgeon General; 2001.
Breslau J, Kendler KS, Su M, Gaxiola-Aguilar S, Kessler RC. Lifetime risk and persistence of psychiatric disorders across ethnic groups in the United States. Psychol Med. 2005;35:317–27.
Waraich P, Goldner EM, Somers JM, Hsu L. Prevalence and incidence studies of mood disorders: a systematic review of the literature. Can J Psychiatry. 2004;49:124–38.
Dunlop DD, Song J, Lyons JS, Manheim LM, Chang RW. Racial/ethnic differences in rates of depression among preretirement adults. Am J Public Health. 2003;93:1945–52.
Kawakami N, Takeshima T, Ono Y, Uda H, Hata Y, Nakane Y, et al. Twelve-month prevalence, severity, and treatment of common mental disorders in communities in Japan: preliminary finding from the World Mental Health Japan Survey 2002–2003. Psychiatry Clin Neurosci 2005; 59:441–52.
Riolo SA, Nguyen TA, Greden JF, King CA. Prevalence of depression by race/ethnicity: findings from the National Health and Nutrition Examination Survey III. Am J Public Health. 2005;95:998–1000.
Straub RH, Dhabhar FS, Bijlsma JW, Cutolo M. How psychological stress via hormones and nerve fibers may exacerbate rheumatoid arthritis. Arthr Rheum. 2005;52:16–26.
Dhabhar FS, McEwen BS. Acute stress enhances while chronic stress suppresses immune function in vivo: a potential role for leukocyte trafficking. Brain Behav Immun. 1997;11:286–306.
Dhabhar FS, McEwen BS. Enhancing versus suppressive effects of stress hormones on skin immune function. Proc Natl Acad Sci USA. 1999;96:1059–64.
Dhabhar FS, McEwen BS. Stress-induced enhancement of antigen-specific cell-mediated immunity. J. Immunol. 1996;156:2608–15.
McEwen BS. Protective and damaging effects of stress mediators: allostasis and allostatic load. N Engl J Med. 1998;338:171–9.
Sephton SE, Sapolsky RM, Kraemer HC, Spiegel D. Diurnal cortisol rhythm as a predictor of breast cancer survival. J Natl Cancer Inst. 2000;92:994–1000.
Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocr Rev. 1984;5:25–44.
Schleimer RP, Claman HN, Oronsky A, editors. Anti-inflammatory steroid action: basic and clinical aspect. San Diego: Academic Press, Inc.; 1989.
Dohrenwend BP, Levav I, Shrout PE, Schwartz S, Naveh G, Link BG, et al. Socioeconomic status and psychiatric disorders: the causation-selection issue. Science. 1992;255:946–52.
Bruce ML, Hoff RA. Social and physical health risk factors for first-onset major depressive disorder in a community sample. Soc Psychiatry Psychiatr Epidemiol. 1994;29:165–71.
Swindle RW, Cronkite RC, Moos RH. Life stressors, social resources, coping, and the 4-year course of unipolar depression. J Abnorm Psychol. 1989;98:468–77.
Simon GE. Social and economic burden of mood disorders. Biol Psychiatry. 2003;54:208–15.
Breslau N, Davis GC. Chronic stress and major depression. Arch Gen Psychiatry. 1986;43:309–14.
Paykel ES, Cooper Z. Life events and social stress. In: Paykel ES, editor. Handbook of affective disorder. New York: Guilford Press; 1992. p. 149–70.
Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: Review and meta-analysis. Am J Psychiatry. 2000;157:1552–62.
Bierut LJ, Heath AC, Bucholz KK, Dinwiddie SH, Madden PA, Statham DJ, et al. Major depressive disorder in a community-based twin sample: are there different genetic and environmental contributions for men and women? Arch Gen Psychiatry. 1999;56:557–63.
Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. The lifetime history of major depression in women. Reliability of diagnosis and heritability. Arch Gen Psychiatry. 1993;50:863–70.
Kendler KS, Gardner CO, Neale MC, Prescott CA. Genetic risk factors for major depression in men and women: similar or different heritabilities and same or partly distinct genes? Psychol Med. 2001;31:605–16.
McGuffin P, Katz R, Rutherford J. Nature, nurture and depression: a twin study. Psychol Med. 1991;21:329–55.
McGuffin P, Katz R, Watkins S, Rutherford J. A hospital-based twin register of the heritability of DSM-IV unipolar depression. Arch Gen Psychiatry. 1996;53:129–36.
Torgersen S. Genetic factors in moderately severe and mild affective disorders. Arch Gen Psychiatry. 1986;43:222–6.
Mendlewicz J, Rainer JD. Adoption study supporting genetic transmission in manic-depressive illness. Nature. 1977;268:326–9.
Cadoret R. Evidence for genetic inheritance of primary affective disorder in adoptees. Am J Psychiatry. 1978;133:463–6.
Wender PH, Kety SS, Rosenthal D, Schulsinger F, Ortmann J, Lunde I. Psychiatric disorders in the biological and adoptive families of adopted individuals with affective disorders. Arch Gen Psychiatry. 1986;43:923–9.
Gershon ES, Weissman MM, Guroff JJ, Prusoff BA, Leckman JF. Validation of criteria for major depression through controlled family study. J Affect Disord. 1986;11:125–31.
Weissman MM, Gershon ES, Kidd KK, Prusoff BA, Leckman JF, Dibble E, et al. Psychiatric disorders in the relatives of probands with affective disorders The Yale University, National Institute of Mental Health Collaborative Study. Arch Gen Psychiatry. 1984;41:13–21.
Maier W, Lichtermann D, Minges J, Heun R, Hallmayer J, Benkert O. Schizoaffective disorder and affective disorders with mood-incongruent psychotic features: keep separate or combine? Evidence from a family study. Am J Psychiatry. 1992;149:1666–73.
Bunney WE, Davis JM. Noradrenaline in depressive reactions. A review. Arch Gen Psychiatry. 1965;13:483–94.
Schildkraut JJ, Gordon EK, Durell J. Catecholamine metabolism in affective disorders. I. Normetanephrine and VMA excretion in depressed patients treated with imipramine. J Psychiatr Res. 1965;3:213–28.
Coppen A. The biochemistry of affective disorders. Br J Psychiatry. 1967;113:1237–64.
Delgado PL, Charney DS, Price LH, Aghajanian GK, Landis H, Heninger GR. Serotonin function and the mechanism of antidepressant action. Reversal of antidepressant-induced remission by rapid depletion of plasma tryptophan. Arch Gen Psychiatry. 1990;47:411–8.
Miller HL, Delgado PL, Salomon RM, Berman R, Krystal JH, Heninger GR, et al. Clinical and biochemical effects of catecholamine depletion on antidepressant-induced remission of depression. Arch Gen Psychiatry. 1996;53:117–28.
Heils A, Teufel A, Petri S, Stöber G, Riederer P, Bengel D, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem. 1996;66:2621–4.
Heils A, Mößner R, Lesch KP. The human serotonin transporter gene polymorphism-basic research and clinical implications. J Neural Transm. 1997;104:1005–14.
Collier DA, Stöber G, Li T, Heils A, Catalano M, Di Bella D, et al. A novel functional polymorphism within the promoter of the serotonin transporter gene: possible role in susceptibility to affective disorders. Mol Psychiatry. 1996;1:453–60.
McKenzie A, Quinn J. A serotonin transporter gene intron 2 polymorphic region, correlated with affective disorders, has allele-dependent differential enhancer-like properties in the mouse embryo. Proc Natl Acad Sci USA. 1999;96:15251–5.
Abbar M, Courtet P, Amadéo S, Caer Y, Mallet J, Baldy-Moulinier MC, et al. Suicidal behaviors and the tryptophan hydroxylase gene. Arch Gen Psychiatry. 1995;52:846–9.
Peroutka SJ. 5-Hydroxytryptamine receptor subtypes. Ann Rev Neurosci. 1988;11:45–60.
Nielsen DA, Goldman D, Virkkunen M, Tokola R, Rawlings R, Linnoila M. Suicidality and 5-hydroxyindoleacetic acid concentration associated with a tryptophan hydroxylase polymorphism. Arch Gen Psychiatry. 1994;51:34–8.
Du L, Bakish D, Hrdina PD. Tryptophan hydroxylase gene 218A/C polymorphism is associated with somatic anxiety in major depressive disorder. J Affect Disord. 2001;65:37–44.
Gizatullin R, Zaboli G, Jönsson EG, Asberg M, Leopardi R. The tryptophan hydroxylase (TPH) 2 gene unlike TPH-1 exhibits no association with stress-induced depression. J Affect Disord. 2008;107:175–9.
Jokela M, Räikkönen K, Lehtimäki T, Rontu R, Keltikangas-Järvinen L. Tryptophan hydroxylase 1 gene (TPH1) moderates the influence of social support on depressive symptoms in adults. J Affect Disord. 2007;100:191–7.
Christenson JG, Dairman W, Udenfriend S. On the identity of DOPA decarboxylase and 5-hydroxytryptophan decarboxylase (immunological titration-aromatic l-amino acid decarboxylase–serotonin– dopamine–norepinephrine). Proc Natl Acad Sci USA. 1972;69:343–7.
Lotta T, Vidgren J, Tilgmann C, Ulmanen I, Melén K, Julkunen I, et al. Kinetics of human soluble and membrane-bound catechol-O-methyltransferase: a revised mechanism and description of the thermolabile variant of the enzyme. Biochemistry. 1995;34:4202–10.
Massat I, Souery D, Del-Favero J, Nothen M, Blackwood D, Muir W, et al. Association between COMT (Val158Met) functional polymorphism and early onset in patients with major depressive disorder in a European multicenter genetic association study. Mol Psychiatry. 2005;10:598–605.
Ohara K, Nagai M, Suzuki Y, Ohara K. Low activity allele of the catechol-O-methyltransferase gene and Japanese unipolar depression. Neuroreport. 1998;9:1305–8.
Hoyer D, Hannon JP, Martin GR. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol Biochem Behav. 2002;71:533–54.
Alex KD, Pehek EA. Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther. 2007;113:296–320.
Pucadyil TJ, Chattopadhyay A. Cholesterol modulates the antagonist-binding function of hippocampal serotonin1A receptors. Biochim Biophys Acta. 2005;1714:35–42.
Dubovsky SL, Thomas M. Serotonergic mechanisms and current and future psychiatric practice. J Clin Psychiatry. 1995;56(Suppl 2):38–48.
Kapur S, Remigton G. Serotonin–Dopamine interaction and its relevance to schizophrenia. Am J Psychiatry. 1996;153:466–76.
Parsey RV, Oquendo MA, Ogden RT, Olvet DM, Simpson N, Huang YY, et al. Altered serotonin 1A binding in major depression: a [carbonyl-C-11] WAY100635 positron emission tomography study. Biol Psychiatry. 2006;59:106–13.
Lemonde S, Turecki G, Bakish D, Du L, Hrdina PD, Bown CD, et al. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci. 2003;23:8788–99.
Arias B, Catalan R, Gasto C, Gutierrez B, Fananas L. 5-HTTLPR polymorphism of the serotonin transporter gene predicts non-remission in major depression patients treated with citalopram in a 12-weeks follow up study. J Clin Psychopharmacol. 2003;23:563–7.
Duman RS, Charney DS. Cell atrophy and loss in major depression. Biol Psychiatry. 1999;45:1083–4.
Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25.
Jiang X, Xu K, Hoberman J, Tian F, Marko AJ, Waheed JF, et al. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacol. 2005;30:1353–61.
Schumacher J, Jamra RA, Becker T, Ohlraun S, Klopp N, Binder EB, et al. Evidence for a relationship between genetic variants at the brain-derived neurotrophic factor (BDNF) locus and major depression. Biol Psychiatry. 2005;58:307–14.
Strauss J, Barr CL, George CJ, King N, Shaikh S, Devlin B, et al. Association study of brain-derived neurotrophic factor in adults with a history of childhood onset mood disorder. Am J Med Genet B Neuropsychiatr Genet. 2004;131B:6–9.
Surtees PG, Wainwright NW, Willis-Owen SA, Sandhu MS, Luben R, Day NE, et al. No association between the BDNF Val66Met polymorphism and mood status in a non-clinical community sample of 7389 older adults. J Psychiatr Res. 2007;41:404–9.
Cohen S, Rosa A, Corsico A, Sterne A, Owen M, Korzsun A, et al. The brain derived neurotrophic factor (BDNF) Val66Met polymorphism and recurrent unipolar depression. Am J Med Genet B Neuropsychiatr Genet. 2004;130B:37–8.
Kim CH, Kim HS, Cubells JF, Kim KS. A previously undescribed intron and extensive 5′ upstream sequence, but not Phox2a-mediated transactivation, are necessary for high level cell type-specific expression of the human norepinephrine transporter gene. J Biol Chem. 1999;274:6507–18.
Meyer J, Wiedemann P, Okladnova O, Brüss M, Staab T, Stöber G, et al. Cloning and functional characterization of the human norepinephrine transporter gene promoter. J Neural Transm. 1998;105:1341–50.
Chang CC, Lu RB, Chen CL, Chu CM, Chang HA, Huang CC, et al. Lack of association between the norepinephrine transporter gene and major depression in a Han Chinese population. J Psychiatry Neurosci. 2007;32:121–8.
Inoue K, Itoh K, Yoshida K, Higuchi H, Kamata M, Takahashi H, et al. No association of the G1287A polymorphism in the norepinephrine transporter gene and susceptibility to major depressive disorder in a Japanese population. Biol Pharm Bull. 2007;30:1996–8.
Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59:11–4.
Kahn RS, Davis KL. New developments in dopamine and schizophrenia. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology, the Fourth Generation of Progress. New York: Raven Press; 1995.
Souery D, Lipp O, Mahieu B, Mendelbaum K, De Bruyn A, De Maertelaer V, et al. Excess of tyrosine hydroxylase restriction fragment length polymorphism homozygosity in unipolar but not bipolar patients: a preliminary report. Biol Psychiatry. 1996;40:305–8.
Lim LC, Gurling H, Curtis D, Brynjolfsson J, Petursson H, Gill M. Linkage between tyrosine hydroxylase gene and affective disorder cannot be excluded in two of six pedigrees. Am J Med Genet. 1993;48:223–8.
Serretti A, Macciardi F, Verga M, Cusin C, Pedrini S, Smeraldi E. Tyrosine hydroxylase gene associated with depressive symptomatology in mood disorder. Am J Med Genet. 1998;81:127–30.
Furlong RA, Rubinsztein JS, Ho L, Walsh C, Coleman TA, Muir WJ, et al. Analysis and metaanalysis of two polymorphisms within the tyrosine hydroxylase gene in bipolar and unipolar affective disorders. Am J Med Genet. 1999;88:88–94.
Sokoloff P, Giros B, Martres MP, Bouthenet ML, Schwartz JC. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–51.
Willner P, Muscat R, Phillips G. The role of dopamine in rewarded behavior: ability, insight, drive or incentive? Pol J Pharmacol Pharm. 1991;43:291–300.
Willner P, Muscat R, Papp M. An animal model of anhedonia. Clin Neuropharmacol. 1992;15:550–1.
Civelli O, Bunzow JR, Grandy DK. Molecular diversity of the dopamine receptors. Annu Rev Pharmacol Toxicol. 1993;33:281–307.
Ross EM, Gilman AG. Biochemical properties of hormone-sensitive adenylate cyclase. Annu Rev Biochem. 1980;49:533–64.
Arinami T, Gao M, Hamaguchi H, Toru M. A functional polymorphism in the promoter region of the dopamine D2 receptor gene is associated with schizophrenia. Hum Mol Genet. 1997;6:577–82.
Jönsson EG, Nöthen MM, Grünhage F, Farde L, Nakashima Y, Propping P, et al. Polymorphisms in the dopamine D2 receptor gene and their relationships to striatal dopamine receptor density of healthy volunteers. Mol Psychiatry. 1999;4:290–6.
Cravchik A, Sibley DR, Gejman PV. Functional analysis of the human D2 dopamine receptor missense variants. J Biol Chem. 1996;271:26013–7.
Pohjalainen T, Rinne JO, Någren K, Lehikoinen P, Anttila K, Syvälahti EK, et al. The A1 allele of the human D2 dopamine receptor gene predicts low D2 receptor availability in healthy volunteers. Mol Psychiatry. 1998;3:256–60.
Jönsson EG, Flyckt L, Burgert E, Crocq MA, Forslund K, Mattila-Evenden M, et al. Dopamine D3 receptor gene Ser9Gly variant and schizophrenia: association study and meta-analysis. Psychiatr Genet. 2003;13:1–12.
Van Tol HH, Wu CM, Guan HC, Ohara K, Bunzow JR, Civelli O, et al. Multiple dopamine D4 receptor variants in the human population. Nature. 1992;358:149–52.
Asghari V, Sanyal S, Buchwaldt S, Paterson A, Jovanovic V, Van Tol H. Modulation of intracellular cyclic AMP levels by different human dopamine D4 receptor variants. J Neurochem. 1995;65:1157–65.
Manki H, Kanba S, Muramatsu T, Higuchi S, Suzuki E, Matsushita S, et al. Dopamine D2, D3 and D4 receptor and transporter gene polymorphisms and mood disorders. J Affect Disord. 1996;40:7–13.
Rees M, Norton N, Jones I, McCandless F, Scourfield J, Holmans P, et al. Association studies of bipolar disorder at the human serotonin transporter gene (hSERT; 5HTT). Mol Psychiatry. 1997;2:398–402.
Frodl T, Meisenzahl EM, Zill P, Baghai T, Rujescu D, Leinsinger G, et al. Reduced hippocampal volumes associated with the long variant of the serotonin transporter polymorphism in major depression. Arch Gen Psychiatry. 2004;61:177–83.
Dannlowski U, Ohrmann P, Bauer J, Deckert J, Hohoff C, Kugel H, et al. 5-HTTLPR biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacol. 2008;33:418–24.
Hoehe MR, Wendel B, Grunewald I, Chiaroni P, Levy N, Morris-Rosendahl D, et al. Serotonin transporter (5-HTT) gene polymorphisms are not associated with susceptibility to mood disorders. Am J Med Genet. 1998;81:1–3.
Frisch A, Postilnick D, Rockah R, Michaelovsky E, Postilnick S, Birman E, et al. Association of unipolar major depressive disorder with genes of the serotonergic and dopaminergic pathways. Mol Psychiatry. 1999;4:389–92.
Geijer T, Frisch A, Persson ML, Wasserman D, Rockah R, Michaelovsky E, et al. Search for association between suicide attempt and serotonergic polymorphisms. Psychiatr Genet. 2000;10:19–26.
Taylor WD, Steffens DC, Payne ME, MacFall JR, Marchuk DA, Svenson IK, et al. Influence of serotonin transporter promoter region polymorphisms on hippocampal volumes in late-life depression. Arch Gen Psychiatry. 2005;62:537–44.
Grünblatt E, Löffler C, Zehetmayer S, Jungwirth S, Tragl KH, Riederer P, et al. Association study of the 5-HTTLPR polymorphism and depression in 75-year-old non-demented subjects from the Vienna Transdanube Aging (VITA) study. J Clin Psychiatry. 2006;67:1373–8.
Bellivier F, Henry C, Szöke A, Schürhoff F, Nosten-Bertrand M, Feingold J, et al. Serotonin transporter gene polymorphisms in patients with unipolar or bipolar depression. Neurosci Lett. 1998;255:143–6.
Furlong RA, Ho L, Walsh C, Rubinsztein JS, Jain S, Paykel ES, et al. Analysis and meta-analysis of two serotonin transporter gene polymorphisms in bipolar and unipolar affective disorders. Am J Med Genet. 1998;81:58–63.
Minov C, Baghai TC, Schüle C, Zwanzger P, Schwarz MJ, Zill P, et al. Serotonin-2A-receptor and -transporter polymorphisms: lack of association in patients with major depression. Neurosci Lett. 2001;303:119–22.
Serretti A, Lilli R, Lorenzi C, Lattuada E, Cusin C, Smeraldi E. Serotonin transporter gene (5-HTTLPR) and major psychoses. Mol Psychiatry. 2002;7:95–9.
Cervilla JA, Rivera M, Molina E, Torres-González F, Bellón JA, Moreno B, et al. The 5-HTTLPR s/s genotype at the serotonin transporter gene (SLC6A4) increases the risk for depression in a large cohort of primary care attendees: the PREDICT-gene study. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:912–7.
Dorado P, Peñas-Lledó EM, González AP, Cáceres MC, Cobaleda J, Llerena A. Increased risk for major depression associated with the short allele of the serotonin transporter promoter region (5-HTTLPR-S) and the CYP2C9*3 allele. Fundam Clin Pharmacol. 2007;21:451–3.
Hickie IB, Naismith SL, Ward PB, Scott EM, Mitchell PB, Schofield PR, et al. Serotonin transporter gene status predicts caudate nucleus but not amygdala or hippocampal volumes in older persons with major depression. J Affect Disord. 2007;98:137–42.
Hauser J, Leszczyńska A, Samochowiec J, Czerski PM, Ostapowicz A, Chlopocka M, et al. Association analysis of the insertion/deletion polymorphism in serotonin transporter gene in patients with affective disorder. Eur Psychiatry. 2003;18:129–32.
Hoefgen B, Schulze TG, Ohlraun S, von Widdern O, Höfels S, Gross M, et al. The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry. 2005;57:247–51.
Kunugi H, Hattori M, Kato T, Tatsumi M, Sakai T, Sasaki T, et al. Serotonin transporter gene polymorphisms: ethnic difference and possible association with bipolar affective disorder. Mol Psychiatry. 1997;2:457–62.
Ohara K, Nagai M, Tani K, Nakamura Y, Ino A, Ohara K. Functional polymorphism of −141C Ins/Del in the dopamine D2 receptor gene promoter and schizophrenia. Psychiatry Res. 1998;81:117–23.
Kim DK, Lim SW, Lee S, Sohn SE, Kim S, Hahn CG, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport. 2000;11:215–9.
Lotrich FE, Pollock BG. Meta-analysis of serotonin transporter polymorphisms and affective disorders. Psychiatr Genet. 2004;14:121–9.
Lasky-Su JA, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of the association between two polymorphisms in the serotonin transporter gene and affective disorders. Am J Med Genet Part B. 2005;133B:110–5.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88.
Mendlewicz J, Massat I, Souery D, Del-Favero J, Oruc L, Nöthen MM, et al. Serotonin transporter 5HTTLPR polymorphism and affective disorders: no evidence of association in a large European multicenter study. Eur J Hum Genet. 2004;12:377–82.
Matullo G, Peluso M, Polidoro S, Guarrera S, Munnia A, Krogh V, et al. Combination of DNA repair gene single nucleotide polymorphisms and increased levels of DNA adducts in a population-based study. Cancer Epidemiol Biomarkers Prev. 2003;12:674–7.
Goode EL, Ulrich CM, Potter JD. Polymorphisms in DNA repair genes and associations with cancer risk. Cancer Epidemiol Biomarkers Prev. 2002;11:1513–30.
Caporaso NE. Why have we failed to find the low penetrance genetic constituents of common cancers? Cancer Epidemiol Biomarkers Prev. 2002;11:1544–9.
Brennan P. Gene-environment interaction and aetiology of cancer: what does it mean and how can we measure it? Carcinogenesis. 2002;23:381–7.
Ogilvie AD, Battersby S, Bubb VJ, Fink G, Harmar AJ, Goodwim GM, et al. Polymorphism in serotonin transporter gene associated with susceptibility to major depressive disorder. Lancet. 1996;347:731–3.
Stöber G, Heils A, Lesch KP. Serotonin transporter gene polymorphism and affective disorder. Lancet. 1996;347:1340–1.
Gutiérrez B, Pintor L, Gastó C, Rosa A, Bertranpetit J, Vieta E, et al. Variability in the serotonin transporter gene and increased risk for major depressive disorder with melancholia. Hum Genet. 1998;103:319–22.
Mellerup E, Bennike B, Bolwig T, Dam H, Hasholt L, Jørgensen MB, et al. Platelet serotonin transporters and the transporter gene in control subjects, unipolar patients and bipolar patients. Acta Psychiatr Scand. 2001;103:229–33.
Collier DA, Arranz MJ, Sham P, Battersby S, Vallada H, Gill P, et al. The serotonin transporter is a potential susceptibility factor for bipolar affective disorder. Neuroreport. 1996;7:1675–9.
Liu W, Gu N, Feng G, Li S, Bai S, Zhang J, et al. Tentative association of the serotonin transporter with schizophrenia and unipolar depression but not with bipolar disorder in Han Chinese. Pharmacogenetics. 1999;9:491–5.
Wacholder S, Chanock S, Garcia-Closas M, El Ghormli L, Rothman N. Assessing the probability that a positive report is false: an approach for molecular epidemiology studies. J Natl Cancer Inst. 2004;96:434–42.
Healey CS, Dunning AM, Teare MD, Chase D, Parker L, Burn J, et al. A common variant in BRCA2 is associated with both breast cancer risk and prenatal viability. Nat Genet. 2000;26:362–4.
Spurdle AB, Hopper JL, Chen X, Dite GS, Cui J, McCredie MR, et al. The BRCA2 372 HH genotype is associated with risk of breast cancer in Australian women under age 60 years. Cancer Epidemiol Biomarkers Prev. 2002;11:413–6.
d’Errico A, Taioli E, Chen X, Vineis P. Genetic metabolic polymorphisms and risk of cancer: a review of the literature. Biomerkers. 1996;1:149–73.
Garte S, Gaspari L, Alexandrie AK, Ambrosone C, Autrup H, Autrup JL, et al. Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev. 2001;10:1239–48.
Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, et al. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–4.
Eley TC, Sugden K, Corsico A, Gregory AM, Sham P, McGuffin P, et al. Gene–environment interaction analysis or serotonin system markers with adolescent depression. Mol Psychiatry. 2004;9:908–15.
Kaufman J, Yang BZ, Douglas-Palumberi H, Houshyar S, Lipschitz D, Krystal JH, et al. Social supports and serotonin transporter gene moderate depression in maltreated children. Proc Natl Acad Sci USA. 2004;101:17316–21.
Kendler KS, Kuhn JW, Vittum J, Prescott CA, Riley B. The interaction of stressful life events and a serotonin transporter polymorphism in the prediction of episodes of major depression: a replication. Arch Gen Psychiatry. 2005;62:529–35.
Grabe HJ, Lange M, Wolff B, Volzke H, Lucht M, Freyberger HJ, et al. Mental and physical distress is modulated by a polymorphism in the 5-HT transporter gene interacting with social stressors and chronic disease burden. Mol Psychiatry. 2005;10:220–4.
Zalsman G, Huang YY, Oquendo MA, Burke AK, Hu XZ, Brent DA, et al. Association of a triallelic serotonin transporter gene promoter region (5-HTTLPR) polymorphism with stressful life events and severity of depression. Am J Psychiatry. 2006;163:1588–93.
Sjöberg RL, Nilsson KW, Nordquist N, Ohrvik J, Leppert J, Lindström L, et al. Development of depression: sex and the interaction between environment and a promoter polymorphism of the serotonin transporter gene. Int J Neuropsychopharmacol. 2006;9:443–9.
Wilhelm K, Mitchell PB, Niven H, Finch A, Wedgwood L, Scimone A, et al. Life events, first depression onset and the serotonin transporter gene. Br J Psychiatry. 2006;188:210–5.
Gillespie NA, Whitfield JB, Williams B, Heath AC, Martin NG. The relationship between stressful life events, the serotonin transporter (5-HTTLPR) genotype and major depression. Psychol Med. 2005;35:101–11.
Surtees PG, Wainwright NW, Willis-Owen SA, Luben R, Day NE, Flint J. Social adversity, the serotonin transporter (5-HTTLPR) polymorphism and major depressive disorder. Biol Psychiatry. 2006;59:224–9.
Brown GW, Harris TO. Depression and the serotonin transporter 5-HTTLPR polymorphism: a review and a hypothesis concerning gene–environment interaction. J Affect Disord. 2008;111:1–12.
Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, et al. Haplotype variation and linkage disequilibrium in 313 human genes. Science. 2001;293:489–93.
Judson R, Stephens JC, Windemuth A. The predictive power of haplotypes in clinical response. Pharmacogenomics. 2000;1:15–26.
Fallin D, Cohen A, Essioux L, Chumakov I, Blumenfeld M, Cohen D, et al. Genetic analysis of case/control data using estimated haplotype frequencies: application to APOE locus variation and Alzheimer’s disease. Genome Res. 2001;11:143–51.
Khoury M, Beaty TH, Cohen BH. Fundamentals of genetic epidemiology. Monographs in epidemiology and biostatistics. New York: NY Oxford University Press; 1993. p. 144–5.
Hoh J, Wille A, Ott J. Trimming, weighting, and grouping SNPs in human case-control association studies. Genome Res. 2001;11:2115–9.
Acknowledgments
The work was presented in part at the 78th Annual Meeting of the Japanese Society for Hygiene, April 2008, Kumamoto. We thank Professor Norito Kawakami (Department of Mental Health, the University of Tokyo Graduate School of Medicine), the former chairman of the Stress Research Group.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kiyohara, C., Yoshimasu, K. Molecular epidemiology of major depressive disorder. Environ Health Prev Med 14, 71–87 (2009). https://doi.org/10.1007/s12199-008-0073-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12199-008-0073-6