
Abstract
Since the Food and Drug Administration approval of imatinib for treatment of chronic myeloid leukemia in 2001, tyrosine kinase inhibitors (TKIs) have become a mainstay in the care of many malignancies. In acute myeloid leukemia (AML), activating mutations in the FMS-like tyrosine kinase 3 (FLT3) gene result in survival and proliferation of leukemic blasts and are associated with adverse prognosis. Therefore, the FLT3 receptor is an appealing target for inhibition. Multiple small molecule TKIs are currently in development for FLT3-mutated AML, and agents are beginning to show promising efficacy. In other malignancies, the development of resistance to TKIs during the course of therapy has proven to be a challenge, and thus far, in clinical trials of FLT3 TKIs, resistance to inhibition represents a significant barrier to successful FLT3 inhibition. Understanding the mechanisms of resistance and overcoming these obstacles to target inhibition will be central to the success of these agents.
Similar content being viewed by others


Introduction
While cure rates for acute myeloid leukemia (AML) have improved over the past four decades, survival remains suboptimal. Five-year survival for patients under 60 years old is only 40 % [1]. The standard of care for most newly diagnosed patients with AML consists of induction chemotherapy with infusional cytarabine and an anthracycline. In recent years, efforts to introduce new combinations of cytotoxic chemotherapy unfortunately have not resulted in any improvement in overall survival [2]. Mutations in the FMS-like tyrosine kinase 3 (FLT3) gene characterize more than 30 % of AML cases. FLT3 internal tandem duplication (ITD) mutations, accounting for approximately 23 % of AML cases, are associated with a particularly poor prognosis [3–5]. The prognostic implications of FLT3/D835 point mutations found at diagnosis, comprising approximately 7 % of cases, are not yet well established [6–8].
FLT3/ITD AML frequently presents with leukocytosis and normal cytogenetics and is more likely to arise de novo rather than out of an antecedent disorder such as myelodysplastic syndrome (MDS) or a myeloproliferative neoplasm. Patients with this disease are usually not cured with conventional chemotherapy. As a result of failures with chemotherapy alone, allogeneic hematopoietic stem cell transplant (HSCT) early during first remission is rapidly becoming the standard of care for FLT3/ITD AML [9–12]. In addition, a number of small molecule FLT3 tyrosine kinase inhibitors (TKIs) are in development for the treatment of FLT3-mutated AML. These agents are being tested in clinical trials in various contexts—in combination with induction chemotherapy, as single agents in the relapsed/refractory setting, combined with hypomethylating agents in the relapsed/refractory population, and as maintenance therapy before or after HSCT.
Small molecule TKIs are now used routinely in the management of many malignancies, including chronic myeloid leukemia (CML), Philadelphia chromosome positive acute lymphoblastic leukemia, gastrointestinal stromal tumor, non-small cell lung cancer, renal cell carcinoma, hepatocellular carcinoma, and sarcoma. Based on recent data, there is significant expectation that FLT3 inhibitors will have clinical benefit in AML. However, the road to clinical development of FLT3 inhibitors has been an unexpectedly difficult one, with both pharmacokinetic and pharmacodynamic obstacles. This review summarizes the large body of data accumulated regarding the relative efficacy and mechanisms of resistance to this therapeutic approach.
FLT3 receptor in AML
The receptor tyrosine kinase (RTK) FLT3 is a member of the so-called “split-kinase domain” or type III receptors, which include PDGFR, KIT, and FMS [13]. This is important to note, because even relatively selective FLT3 TKIs will routinely have inhibitory activity against some or all of these other RTKs [14]. FLT3 is normally only expressed in primitive hematopoietic precursors within the bone marrow [15]. Activating mutations of FLT3 in AML patients were first reported in 1996, and even in that small series of patients, it was clear that these mutations conferred a poor prognosis [5]. There are two broad categories of activating mutations. The most common abnormalities, occurring in approximately 23 % of newly diagnosed de novo AML, are the FLT3/ITD mutations [6]. These mutations consist of in-frame tandem duplications of coding sequence, almost always encompassing arginine residue 595 within the juxtamembrane domain [16]. The juxtamembrane domain of RTKs often has an autoinhibitory function [17]. Disruption of its secondary structure by mutation frequently results in constitutive activation of the tyrosine kinase [18]. FLT3/ITD mutations vary in length from patient to patient, ranging from three to more than 400 base pairs [19]. Longer mutations, by their nature, result in the duplicated segment starting in the kinase domain. These kinase domain insertions may result in somewhat different biologic effects, and some groups have reported an association with even worse outcomes [20–23]. Whether ITD length impacts the efficacy of FLT3 TKIs is not known. The other major category of FLT3-activating mutation consists of point mutations within the tyrosine kinase domain (TKD). Most commonly, the activation loop at residue aspartate 835 (D835), although several variants at nearby residues have also been reported. The D835 residue can be replaced by a number of amino acids. These mutations are less common than the ITD mutations and do not appear to have the same prognostic impact. However, they are quite important with regard to therapy using FLT3 TKIs, because many of the newer generation FLT3 inhibitors have little or no activity against several, or even most, of the D835 variants. As will be detailed below, D835 mutations occurring in the context of a pre-existing ITD mutation are a common mechanism of resistance to monotherapy with a FLT3 inhibitor.
Over-expression of the wild-type FLT3 receptor is another mechanism of constitutive activation, and, although relatively uncommon in AML, may represent another opportunity for therapy [24]. Most FLT3 TKIs have less activity against the ligand-activated wild-type FLT3 as compared to ITD-mutated receptors. This difference in activity must be considered when designing FLT3 TKI clinical trials.
FMS-like tyrosine kinase 3 has now been well validated as a therapeutic target in AML. While the FLT3/ITD abnormality does not appear to be an initiating mutation, it appears to be one of the most important cooperating mutations in the development of the disease [25]. In patients with FLT3/ITD AML receiving the TKI quizartinib, characteristic and reproducible point mutations often arise at two particular amino acid residues, conferring resistance to therapy with this drug [26]. This discovery of an adaptive resistance mechanism to FLT3 inhibition is direct evidence that the ITD represents a driver mutation in AML. However, the situation with FLT3/ITD AML is much more complex than a simple case of an oncogene-addicted malignant cell. Genome-wide sequencing studies of diagnostic and relapsed AML samples suggest that at presentation an AML cell population is made up of several clonal sub-types, sharing a common mutational ancestry, but each with a unique complement of initiating mutations. At relapse, a dominant clone is more likely to emerge. This may directly relate to FLT3 TKIs because selective FLT3 inhibition rarely induces a cytotoxic effect in vitro in diagnostic FLT3/ITD AML samples, while samples collected at relapse (which tend to have a higher mutant allelic burden) are invariably much more responsive [27]. This suggests that at relapse a FLT3-addicted clone dominates the leukemia cell population.
It is apparent that a number of factors will influence any response to FLT3 inhibitors. These disease features may ultimately include mutant allelic burden, disease status (newly diagnosed vs. relapsed AML), mutation type, and mutation location. Given the heterogeneity of FLT3-mutated AML, it is therefore not surprising that, while these mutations were discovered almost 20 years ago, no FLT3 inhibitor has yet been approved for the treatment of the disease.
Efficacy
Multiple compounds have shown activity as single agents. Clinical activity in trials is measured by overall survival, event-free survival (EFS), response rates, and/or blast reduction. Many clinical trials have also included assessment of the degree of FLT3 inhibition, usually via the plasma inhibitory activity (PIA) assay [28]. In this surrogate assay, a cell line expressing the FLT3/ITD mutation is incubated with the patient’s plasma, and an immunoblot is performed to identify phospho-FLT3 (P-FLT3), the activated form of FLT3. The quantity of P-FLT3 present can be measured by densitometry and expressed as a proportion of the baseline level of P-FLT3. Inhibition to <15 % of baseline is often considered optimal FLT3 inhibition [28–30].
The first wave of FLT3 inhibitors were TKIs that were originally developed for treatment of solid tumors. These drugs were initially designed to inhibit other kinases but were incidentally found to have activity against FLT3. By their very nature, therefore, these drugs inhibited multiple kinases along with FLT3, and with the resultant off-target effects, they often incurred significant toxicity. However, there are now newer agents, such as PLX3397 and quizartinib, that have been developed for the specific purpose of treating patients with FLT3/ITD AML.
Efficacy of FLT3 inhibitors as single agents
Tandutinib
Tandutinib is a relatively selective inhibitor of the type III RTKs, including FLT3. A phase 1 trial examined the use of tandutinib in 40 patients with AML or high-risk MDS [31]. Dose-limiting toxicity was reversible muscular weakness or fatigue. Two of the five evaluable patients with FLT3/ITD mutations, treated at 525 mg and 700 mg twice daily, showed evidence of antileukemic activity, with decreases in both peripheral and bone marrow blasts. In a phase 2 study, 20 patients with FLT3/ITD AML who were refractory, relapsed, or ineligible for induction chemotherapy, were administered tandutinib [32]. Of 15 evaluable patients, two patients had stable disease, and six patients had a decrease in peripheral blood and bone marrow blasts.
Sorafenib
Sorafenib is a TKI with activity against several different kinases, including RAF kinase, VEGFR, c-KIT, and FLT3. Like most FLT3 TKIs, it has much greater activity against the ITD mutation compared with wild-type FLT3 and has little activity against FLT3 with most TKD mutations. Three phase 1 studies have examined sorafenib in AML. In the first of these trials, 16 patients with relapsed or refractory disease were treated with sorafenib at doses ranging from 200 to 400 mg by mouth twice daily [33]. All six patients with FLT3/ITD mutations but no TKD mutation experienced clinical responses. Not surprisingly, three patients with TKD mutations did not exhibit clinical responses. Three of the seven patients with wild-type FLT3 had modest reductions in peripheral blast counts. A second phase 1 study conducted in Canada randomized patients with relapsed or refractory AML and patients >65 with MDS or secondary AML to multiple dose schedules of sorafenib [34]. A dose of 400 mg twice daily was not well tolerated on this study. One patient with the FLT3/ITD mutation exhibited a complete response (CR) to sorafenib. A third phase 1 study revealed that 400 mg by mouth twice daily for days 1 to 21 of a 28 day cycle was the maximum tolerated dose of sorafenib [35]. No patients met criteria for partial response (PR) or CR. 11 of 15 patients exhibited stable disease as the best response. However, in this study, only two patients were positive for the FLT3/ITD mutation.
A report of compassionate use sorafenib in six patients with refractory or relapsed FLT3/ITD AML revealed that all six patients exhibited clinical responses [36]. In a second study, 16 patients with relapsed FLT3/ITD AML following bone marrow transplant received either sorafenib or sorafenib plus chemotherapy. Reduction in peripheral blasts was seen in 80 % of cases, and three patients experienced PRs [37]. The use of sorafenib in the post-transplant setting was further examined by Metzelder et al. [38]. This study evaluated sorafenib use in 65 patients with FLT3/ITD AML, 63 of whom had relapsed or refractory leukemia. Twenty-nine patients had relapsed following HSCT. Sixty-four patients experienced at least a hematologic response to sorafenib. Patients without prior allogeneic HSCT developed resistance to sorafenib significantly earlier than those with prior HSCT. Three patients had sustained responses more than 900 days, and all of these sustained remissions occurred in the HSCT cohort. The authors concluded that not only does sorafenib monotherapy have significant activity in FLT3/ITD AML, but it may also synergize with allogeneic immune effects to induce durable remissions.
Sunitinib
Sunitinib is an oral multi-targeted kinase inhibitor with activity against a large array of kinases, including FLT3, PDGFR, VEGFR, and c-KIT. It appears to be active against some TKD mutations, and seems to have greater than usual activity against wild-type FLT3 compared to other TKIs [39]. Sunitinib was shown in preclinical studies to have activity in a subcutaneous tumor xenograft model and a bone marrow engraftment model [40]. Two phase 1 studies have examined the use of sunitinib in FLT3/ITD AML. The first of these studies enrolled 29 patients with relapsed or refractory AML or who were considered unfit for standard therapy [41]. These patients received sunitinib at escalating doses. Five patients had FLT3 mutations. Significant FLT3 inhibition in vivo was seen in >50 % of patients receiving 200 mg and higher doses of sunitinib. Another Phase 1 examined 15 AML patients receiving sunitinib for 4-week cycles followed by either a 2- or 1-week break [42]. A dose of 50 mg daily was tolerated but was found to be the maximum tolerated dose. The four patients on the study with FLT3 mutations experienced morphologic or partial responses. Two out of 10 evaluable patients with wild-type FLT3 experienced responses.
Midostaurin
Midostaurin is a multi-targeted indolocarbazole with activity against PKC-alpha, VEGFR, KIT, PDGFR, and FLT3. Of note, midostaurin is equally active against ITD-mutated and TKD-mutated FLT3. In a phase 2 study, 20 patients with relapsed/refractory FLT3-mutated AML or high-grade MDS were treated with midostaurin 75 mg by mouth three times daily [43]. The peripheral blast count decreased by at least 50 % in 14 patients, and the bone marrow blast count decreased by at least 50 % in six patients. Two patients exhibited bone marrow blast reduction to <5 % of baseline. Two patients suffered fatal pulmonary events of unclear etiology. A subset of patients from this trial was analyzed for in vivo FLT3 inhibition by means of the PIA assay, and eight of ten patients achieved substantial inhibition of FLT3. Six of these eight patients achieved clinical responses. Importantly, no responses were observed in patients with PIA activity >15 % baseline.
In a phase 2b study, 60 patients with FLT3 wild-type AML and 35 patients with FLT3-mutated AML were randomly assigned to receive midostaurin at a dose of 50 or 100 mg twice daily [44]. Seventy-one percent of patients with mutated FLT3 achieved a reduction in peripheral blasts or bone marrow blasts by 50 % or greater, compared to 42 % of patients with wild-type FLT3.
Lestaurtinib
Lestaurtinib is an indolocarbazole inhibitor with activity against a broad spectrum of kinases, including FLT3. Like midostaurin, it is active against both FLT3/ITD and FLT3/TKD mutants. In preclinical studies, lestaurtinib was shown to inhibit wild-type and mutant FLT3 in vitro, and there was a high degree of correlation between FLT3 inhibition and cytotoxicity [45]. Lestaurtinib also inhibited FLT3 activation in vivo and prolonged survival in a mouse model of FLT3/ITD AML. Subsequently, in a phase 1/2 trial, lestaurtinib was used as salvage therapy for 14 patients with refractory, relapsed, or poor-risk FLT3-mutated AML [29]. The drug was well-tolerated, and toxicities included mild nausea, mild emesis, mild weakness, and fatigue. Five patients exhibited significant reductions in peripheral blood blasts by >50 %. One patient had a complete response, as defined by bone marrow blast reduction to <5 %. Clinical responses correlated with sustained FLT3 inhibition as assessed by the PIA assay.
This initial study was followed by a phase 2 study of lestaurtinib as monotherapy in untreated older patients with AML not considered candidates for induction chemotherapy [46]. Patients with wild-type and mutant FLT3 AML were included. Patients received lestaurtinib orally for 8 weeks, initially at a dose of 60 mg twice daily, escalating to 80 mg twice daily, and the drug was generally well tolerated. Toxicities included nausea, emesis, diarrhea, constipation, and rise in alkaline phosphatase levels. Three of five patients with mutated FLT3 exhibited hematologic responses. Five of 22 wild-type FLT3 patients achieved a partial or complete bone marrow response. As seen in the phase 1/2 study, clinical responses correlated with sustained FLT3 inhibition as assessed by PIA.
KW-2449
In contrast to the numerous multi-kinase inhibitors that have been adopted as FLT3 inhibitors, KW-2449 was developed specifically to target FLT3. In the phase 1 study of KW-2449, 37 patients were enrolled. Eight patients showed >50 % reduction in peripheral blood blasts. Five of eight responders had FLT3/ITD mutations. Correlative studies suggested that effective, sustained FLT3 inhibition was not achieved in most patients at the maximum tolerated dose. FLT3 activity was down-regulated at 2 and 4 h postdose but was up-regulated at 12 h [35]. Development of this drug was terminated for this reason. While this was a disappointment, the trial was a demonstration of the utility of correlative studies in this field. Maximum, sustained inhibition of FLT3 signaling appears to be essential to achieving any sort of clinical response.
Quizartinib (AC220)
Like KW-2449, quizartinib was designed specifically as a FLT3 inhibitor, and work over the past few years has revealed this drug to be an exceptionally potent and selective agent [47]. In a phase 1 study, quizartinib was administered orally at escalating doses of 12–450 mg/day to 76 patients with relapsed/refractory AML (with a median of 3 prior therapies) enrolled irrespective of FLT3/ITD mutational status. Of 17 FLT3/ITD AML patients, 9 responded (53 %). Composite complete response (CRc) among this group was 24 %, and there were 5 PRs, as well. FLT3 phosphorylation was completely inhibited at virtually all doses tested in the PIA assay.
Following this, a Phase 2 study has recently completed accrual, and preliminary results have been presented in abstract form [48, 49]. In this study, there were two cohorts of patients with FLT3/ITD AML. The first cohort, consisting of 92 patients aged 60 and over who had relapsed or were refractory to front-line therapy, showed an overall response rate (CRc + PR) of 72 %. The second cohort consisted of 99 FLT3/ITD AML patients who had relapsed or were refractory to two lines of therapy or following an allogeneic transplant. These patients had a median age of 50, and the overall response rate (CRc + PR) was 68 %. Significantly, 34 % of these patients were bridged to an allogeneic transplant as a result of this therapy, with a number of long-term survivors. Clinical study of quizartinib is ongoing.
Crenolanib
Crenolanib was originally developed as an inhibitor of PDGFR. It has promising activity against FLT3/ITD and FLT3/TKD mutant isoforms in in vitro binding studies, cell line and murine leukemia models, as well as in primary human AML cells [50]. This includes activity against most TKD mutations arising in the setting of prior FLT3 TKI therapy. A phase I and phase Ib study established the safety of crenolanib in 59 and 48 solid tumor patients, respectively [51, 52]. The efficacy of crenolanib as a FLT3 TKI is currently being investigated in two phase 2 clinical trials of AML patients, one of which is specifically enrolling patients that have progressed on prior FLT3 TKI therapy. Now that agents are arising that have activity against the TKD mutation, the model for FLT3/ITD AML is beginning to approximate the CML model, where specific resistance-conferring mutations arise during therapy with one TKI, necessitating a change to a different inhibitor [53].
Efficacy of FLT3 inhibitors in combination with chemotherapy
In the past two decades, one of the most significant advances in the care of leukemia patients has been the addition of all-trans retinoic acid to high dose anthracyclines to treat patients with acute promyelocytic leukemia (APL). In this case, the addition of a targeted agent to cytotoxic chemotherapy has been responsible for a significant increase in the number of patients who are cured [54]. Following this model of adding a targeted agent to standard treatment regimens, multiple trials have combined a FLT3 inhibitor with chemotherapy to learn whether such combinations may improve outcomes for patients.
Sorafenib
In a phase 1/2 trial, sorafenib 400 mg by mouth twice daily on days 1–7 was combined with a standard regimen of cytarabine and idarubicin in patients under the age of 65 [55]. Ten patients were treated on the phase 1 component of the study, and 51 patients were evaluated as part of the phase 2 component. Of these 51 patients, 75 % achieved complete remission, including 14 of 15 patients with mutated FLT3 (the patient who did not achieve CR achieved CRp, CR with insufficient platelet recovery). Results of the PIA assay indicated an excellent degree of in vivo inhibition of FLT3 in these patients after 1 week of treatment with sorafenib. In another phase 1/2 trial, sorafenib was combined with low dose cytarabine in 21 patients with AML or MDS [56]. In this study, only three patients had FLT3 mutations. Response rate among eligible patients was 10 %.
In a German phase 2 study, 197 AML patients were randomized to receive up to two cycles of standard 7 + 3 induction chemotherapy followed by two cycles of consolidation with intermediate dose cytarabine. Before the start of treatment, patients were randomly assigned to receive either placebo or sorafenib. Sorafenib was administered at a dose of 400 mg twice daily between cycles and after chemotherapy for up to 1 year after induction. There was no difference in EFS or overall survival (OS) between the groups. Among the 28 patients in the study with the FLT3/ITD mutation, there was no difference in EFS or OS [57]. One limitation of this study was the small number of patients with the FLT3/ITD mutation.
Sunitinib
A phase 2 study examined the feasibility of combining sunitinib with standard induction chemotherapy in newly diagnosed FLT3-mutated AML patients ≥60 [58]. CR rate was 59 % in all patients, 53 % in patients with FLT3/ITD AML, and 71 % in patients with FLT3/TKD mutations. The maximum tolerated dose was found to be 25 mg daily when patients took sunitinib on days 1 through 7 of chemotherapy and continuously during maintenance.
Midostaurin
A phase 1b study examined three different dose schedules of midostaurin combined with daunorubicin and cytarabine induction followed by high dose cytarabine maintenance in patients with newly diagnosed AML [59]. A 50 mg twice daily dosing schedule (either on days 1–7 and days 15–21 or days 8–21) was tolerated better than 100 mg twice daily regimens. Of the 40 patients receiving 50 mg twice daily dosing, 45 % discontinued therapy. In this cohort, there was an 80 % CR rate. 12 of 13 patients with FLT3 mutations achieved CR. This trial served as a pilot for a randomized phase 3 study of conventional chemotherapy and midostaurin for newly diagnosed adults with FLT3-mutated AML. This large trial (“RATIFY”) has recently completed accrual, but results have not yet been reported.
Lestaurtinib
In a randomized phase 2 trial, 224 FLT3-mutated patients in first relapse were assigned to receive chemotherapy or chemotherapy plus lestaurtinib [30]. Lestaurtinib was initiated 2 days following the end of chemotherapy. There was no difference in overall survival between the two arms of this study. Only 27 % of patients receiving lestaurtinib achieved sustained, adequate FLT3 inhibition, and this suboptimal FLT3 inhibition was thought due to the complex pharmacokinetics of the drug. There was a high degree of correlation between the strength of FLT3 inhibition and response rates. This relationship is encouraging for future development of FLT3 TKIs. In the UK, a phase 3 study that incorporates lestaurtinib into therapy for FLT3-mutated patients has recently completed accrual. Results have not been reported yet.
Resistance
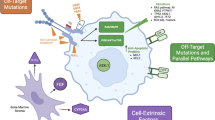
Resistance to a targeted therapy, resulting in a primary lack of efficacy or loss of prior efficacy, can be derived from multiple pharmacokinetic (extrinsic) and pharmacodynamic (intrinsic) factors. In the instance of FLT3 inhibition, we can divide these factors into extrinsic, receptor-intrinsic, and cell-intrinsic factors (Fig. 1).

Mechanisms of resistance to FLT3 inhibition may be categorized as extrinsic, receptor-intrinsic, and cell-intrinsic
As is the case in the treatment of many malignancies, resistance to therapy is a major obstacle in FLT3-mutated AML. For example, as noted above, some of the early phase FLT3 inhibitor trials have shown relatively low efficacy rates. The initial failures of FLT3 inhibitors may in part be due to the fact that many trials were performed in patients without regard to FLT3 mutational status. Moreover, FLT3 inhibition has been suboptimal in patients on several clinical trials [29, 30, 43, 60, 61]. There are myriad established and postulated mechanisms of resistance to FLT3 inhibitors. Some individuals receiving TKIs for FLT3-mutated AML are resistant to therapy from the outset, and others develop resistance during the course of treatment [60]. These two groups of patients have sometimes been described as having “primary resistance” and “secondary resistance” [60].
Extrinsic mechanisms of resistance
Pharmacokinetics can significantly impact the efficacy of FLT3 inhibitors. Insufficient plasma drug levels have been a recurrent impediment in FLT3 inhibitor trials [30, 35]. Hepatic metabolism, complicated by drug interactions, can further make it difficult to maintain consistent plasma drug levels. For instance, midostaurin is metabolized in the liver by CYP3A4 into two metabolites. Either midostaurin or its metabolites may induce CYP3A4, thereby changing the levels of the drug over time [44, 62]. Patients on medications that induce or inhibit CYP3A4 have been excluded from trials of midostaurin [44]. Likewise, it has been shown that medications, including azoles, can alter the metabolism of FLT3 inhibitors such as lestaurtinib [63]. While drug interactions can sometimes potentiate the efficacy of FLT3 TKIs, they can also increase the side effects of a drug and/or make dose-finding challenging in trials.
Additional pharmacokinetic obstacles exist. Protein binding, for instance, can be a major issue. Some FLT3 inhibitors, which are generally hydrophobic and relatively insoluble, are largely bound to alpha-1 acid glycoprotein (AGP). This protein binding often results in diminished free levels of the drugs. Moreover, as shown in the phase 3 lestaurtinib trial, AGP levels can rise with AML therapy [30, 64]. As such, more lestaurtinib can then be bound by AGP, thereby reducing the levels of active lestaurtinib. In fact, lestaurtinib can displace anthracyclines from serum proteins, potentially increasing toxicity of the drug [65]. Expression of ATP-binding cassette multi-drug transporters may be responsible for reduction of intracellular levels of FLT3 TKIs [66]. Finally, it is possible that the bone marrow microenvironment may provide a protective niche for cancer cells, making these cells less susceptible to drugs [67, 68]. FLT3 inhibitors appear to have a different effect upon bone marrow leukemia cells than circulating blasts. While quizartinib will induce cell apoptosis in circulating blasts, this agent promotes differentiation of bone marrow blasts [69].
Another proposed mechanism of resistance involves the cognate ligand for the FLT3 receptor, FLT3 ligand (FL). It is known that FL levels rise when chemotherapy is administered to AML patients [70, 71]. For example, lestaurtinib as single agent therapy was effective at inhibiting FLT3 in vivo, but when given immediately following chemotherapy was less effective at inhibiting the target [70]. Since FL concentrations increased markedly with successive courses of chemotherapy, it was postulated that high FL levels were interfering with FLT3 inhibition. To test this hypothesis, investigators examined FLT3 inhibition in vitro in the presence and absence of FL. At the concentrations observed in AML patients, exogenous FL attenuated FLT3 inhibition for each of five different FLT3 TKIs.
Since FL levels appear to correlate with therapy and aplasia, it is unknown whether high FL concentrations are an effect of the aplasia or, on the other hand, a direct effect of particular treatments. Since chemotherapy promotes elevations in FL concentrations and since high FL levels seem to interfere with FLT3 inhibition, the FL surge with chemotherapy presents a significant challenge for FLT3 inhibition [72]. It is possible that this resistance mechanism can be overcome by more potent FLT3 TKIs. It is also possible that less cytotoxic treatment paradigms may lead to lower levels of FL, thereby creating a more favorable environment for FLT3 inhibition [73].
Receptor-intrinsic mechanisms of resistance
In vitro studies initially suggested that cytotoxicity with FLT3 inhibition was greater in patients with mutated FLT3 than wild-type [74]. Results of multiple early phase clinical trials have corroborated the hypothesis that patients with wild-type FLT3 are less likely to respond to TKIs than patients with a mutated FLT3 receptor [29, 41, 43]. Furthermore, while many FLT3 inhibitors have activity against leukemic cells harboring the FLT3/ITD mutation, cells with particular TKD mutations are inherently resistant to specific agents. Among FLT3 TKIs, the spectrum of activity against the assortment of common TKD mutations is highly variable. For instance, tandutinib can inhibit FLT3 in leukemic cells with the ITD and I836del mutations in comparatively low micromolar concentrations, but even with 20-fold concentrations cannot inhibit FLT3 receptors harboring the D835V or D835Y mutations [75]. Variations in the inhibitory properties of FLT3 TKIs against different TKD mutations have been shown in vitro with other agents such as midostaurin, as well [76]. As noted earlier, patients with mutations at the D835 locus will usually have a reduced response to agents such as sorafenib and quizartinib, depending on the amino acid residue that is substituted for the aspartate [26].
In addition to existing occasionally in the setting of newly diagnosed AML, TKD point mutations in the FLT3 gene, such as mutations at the D835 and F691L loci, frequently arise in response to TKI exposure, both in vitro and in vivo [26, 77–79]. In CML, point mutations can prevent imatinib from accessing the ATP binding pocket of the BCR-ABL kinase. Likewise, in FLT3-mutated AML, such mutations can confer resistance to FLT3 TKIs. Resistance patterns vary from TKI to TKI, with some groups of inhibitors generating non-overlapping resistance [80]. Resistance-conferring mutations are common in the field of TKI therapy, both in treatment of solid tumors and CML. In CML, while dasatinib can overcome most forms of imatinib resistance, the T315I mutation can confer resistance to imatinib, dasatinib, and nilotinib [80, 81]. The newer TKI ponatinib is now available to treat patients with this point mutation [82]. In CML, the current practice is to change TKIs when new mutations are recognized in a patient. Similarly, in FLT3-mutated AML, the varying resistance patterns of different inhibitors may give rise to a situation where either switching of therapy is necessitated by acquired mutations or combinations of therapy are necessary to avoid development of resistance [53].
The sensitivity of a tumor to TKI therapy commonly relies on the presence of an activating mutation in the receptor. Thus far, this seems to hold true in the case of FLT3 and AML. While there has been some in vitro evidence that over-expression of wild-type FLT3 may result in sensitivity to FLT3 inhibition [24], there have been only anecdotal reports of this phenomenon occurring clinically [83]. It is known that the FLT3 receptor can be phosphorylated in wild-type leukemic blasts from AML patients; however, the implications of this finding for therapy are unknown [84]. Regardless of whether overexpression of wild-type FLT3 may play a role in susceptibility to TKIs, modification of gene expression may create an obstacle to treatment. For example, it has been observed that up-regulation of FLT3 occurs in the context of FLT3 inhibition with midostaurin [85]. This type of feedback loop, whereby inhibition promotes an increase in the amount of the receptor, may represent a resistance mechanism to effective inhibition.
Finally, in AML patients with ITD mutations, the characteristics of the ITD may play a role in determining response to therapy. For example, investigators have suggested that the location of the ITD in the TKD domain (as opposed to the juxtamembrane domain) correlates with poor response to chemotherapy [20, 86]. In addition, there is in vitro data indicating that FLT3/ITD blasts that have the ITD inserted into the TKD domain are particularly resistant to FLT3 inhibition [86]. This resistance is thought to be mediated by up-regulation of the anti-apoptotic protein MCL-1. It has also been suggested that long ITD lengths correlate with inferior survival compared to shorter duplication lengths [87]. This relationship between ITD size and prognosis may result from the fact that longer duplications are more likely to involve the TKD [23].
Cell-intrinsic mechanisms of resistance
Primary resistance to FLT3 inhibition can occur through persistent activation of pathways downstream of the FLT3 receptor. For example, the STAT5 and MAPK pathways can remain active even when FLT3 is successfully inhibited with a TKI [83]. Stromal factors can lead to activation of the MAPK pathway [88]. While the mechanism of STAT5 and MAPK activation in such cases are not known, this finding suggests that the presence of redundant signaling cascades may render leukemic cells independent of FLT3. It seems logical that other kinases on the cell surface may be activating these pathways.
Mutations in genes other than FLT3 can also generate obstacles to therapy. This parallels the situation in CML, where overexpression of genes in pathways other than BCR-ABL and/or mutations in those pathways can give rise to resistance [89–91]. In the case of FLT3-mutated AML, some such mutations can occur in genes downstream of the FLT3 receptor. Patients who achieve remissions from FLT3-mutated AML can relapse with Ras mutations [92]. It has also been shown that exposure to FLT3 inhibition can lead to Ras mutations [93]. In addition, mutations in Cbl can interfere with degradation of the FLT3 receptor, thereby leading to increased FLT3 activity [94]. Mutations or expression modification of genes regulating apoptosis, such as BCL2, MCL-1, and NF-κB, can render leukemic cells independent of FLT3 signaling [86, 95–97]. In an in vitro model, midostaurin resistance appeared to be in part governed by up-regulation of anti-apoptotic signals and down-regulation of pro-apoptotic pathways [98].
Multiple clinical studies have shown that the FLT3 mutational status of leukemic cells is unstable [92, 99–104]. The ratio of mutant to wild-type FLT3 varies among patients and correlates with prognosis [104]. In addition, in any one patient, the FLT3 allelic burden can be different at diagnosis and at relapse. As AML patients are treated with chemotherapy or FLT3 TKIs, clonal evolution—including changes in the allelic burden—can take place [25]. At one extreme end of the spectrum, patients who initially present with FLT3-mutated AML occasionally relapse with wild-type FLT3 AML, in which the leukemia cells do not appear to be dependent upon FLT3 signaling [105]. This phenomenon is uncommon, and usually occurs in the setting of a low FLT3 allelic burden at diagnosis and suggests that the small FLT3-mutated clone at diagnosis is not the dominant clone in the patient’s leukemia. Evolution away from a FLT3-mutated dominant clone may comprise a mechanism of resistance to therapy. On the other extreme end of the spectrum, wild-type FLT3 AML can relapse as FLT3-mutated disease. In vitro, FLT3 inhibitors are more likely to have a cytotoxic effect in the setting of relapsed disease and in the context of a high allelic burden [27]. The selection of a dominant AML clone that is characterized by FLT3-addicted disease is probably associated with TKI susceptibility.
Conclusions
FLT3 inhibitors are beginning to show promise in clinical trials, and there is currently great enthusiasm that small molecule TKIs will transform the care of a large number of AML patients. Weak inhibition and transient inhibition have been major obstacles to the development of these agents. We are now equipped with an increasing number of tools that may aid in overcoming barriers to optimal FLT3 inhibition. The PIA assay, for example, allows us to assess whether FLT3 inhibitors are hitting the intended target.
New treatment strategies may allow us to circumvent or surmount resistance to FLT3 TKIs. First, multiple agents are in development that target not only the FLT3/ITD mutation, but also and acquired TKD point mutations. Second, more potent and selective FLT3 inhibitors, such as quizartinib, seem to have better pharmacokinetic properties than early FLT3 TKIs. Finally, an intriguing strategy might be to use less intensive chemotherapy regimens that do not promote high concentrations of FL.
FLT3 inhibition is part of a larger, evolving pattern that has already led to some successes in oncology—namely, the identification of a driver mutation as a molecular target, the development of an agent with activity against this target, the advent of biomarkers to determine whether the drug is having the intended effect, and the selection of patients for use of this agent. In the field of leukemia, APL provides a paradigm for combining a targeted agent with cytotoxic chemotherapy. The combination of a TKI with chemotherapy is a logical approach in FLT3-mutated AML. Novel approaches to overcoming intrinsic and acquired resistance to therapy will likely continue to aid the development of FLT3 inhibitors.
References
Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051–62.
Buchner T, Schlenk RF, Schaich M, Döhner K, Krahl R, Krauter J, et al., et al. Acute Myeloid Leukemia (AML): different treatment strategies versus a common standard arm—combined prospective analysis by the German AML Intergroup. J Clin Oncol. 2012;30:3604–10.
Marcucci G, Maharry K, Whitman SP, Vukosavljevic T, Paschka P, Langer C, et al. High expression levels of the ETS-related gene, ERG, predict adverse outcome and improve molecular risk-based classification of cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B Study. J Clin Oncol. 2007;25:3337–43.
Fröhling S, Schlenk RF, Breitruck J, Benner A, Kreitmeier S, Tobis K, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100:4372–80.
Nakao M, Yokota S, Iwai T, Kaneko H, Horiike S, Kashima K, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–8.
Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17:1738–52.
Whitman SP, Ruppert AS, Radmacher MD, Mrozek K, Paschka P, Langer C, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111:1552–9.
Wang W, Wang XQ, Xu XP, Lin GW. Prevalence and prognostic significance of FLT3 gene mutations in patients with acute leukaemia: analysis of patients from the Shanghai Leukaemia Co-operative Group. J Int Med Res. 2010;38:432–42.
Schlenk RF, Döhner K, Krauter J, Fröhling S, Corbacioglu A, Bullinger L, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–18.
Kayser S, Döhner K, Krauter J, Casper J, Horst HA, Held G, et al. Impact of allogeneic transplantation from matched related and unrelated donors on clinical outcome in younger adult AML patients with FLT3 internal tandem duplications. Blood (ASH Annual Meeting Abstracts). 2010; 116:Abstract 909.
Bornhauser M, Illmer T, Schaich M, Soucek S, Ehninger G, Thiede C. Improved outcome after stem-cell transplantation in FLT3/ITD-positive AML. Blood. 2007;109:2264–5.
DeZern AE, Sung A, Kim S, Smith BD, Karp JE, Gore SD, et al. Role of allogeneic transplantation for FLT3/ITD acute myeloid leukemia: outcomes from 133 consecutive newly diagnosed patients from a single institution. Biol Blood Marrow Transplant. 2011;17:1404–9.
Plowman GD, Sudarsanam S, Bingham J, Whyte D, Hunter T. The protein kinases of Caenorhabditis elegans: a model for signal transduction in multicellular organisms. Proc Natl Acad Sci. 1999;96:13603–10.
Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotech. 2008;26:127–32.
Rosnet O, Bühring HJ, de Lapeyrière O, Beslu N, Lavagna C, Marchetto S, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta Haematol. 1996;95:218–23.
Vempati S, Reindl C, Kaza SK, Kern R, Malamoussi T, Dugas M, et al. Arginine 595 is duplicated in patients with acute leukemias carrying internal tandem duplications of FLT3 and modulates its transforming potential. Blood. 2007;110:686–94.
Hubbard SR. Juxtamembrane autoinhibition in receptor tyrosine kinases. Nat Rev Mol Cell Biol. 2004;5:464–71.
Griffith J, Black J, Faerman C, Swenson L, Wynn M, Lu F, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13:169–78.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66.
Kayser S, Schlenk RF, Londono MC, Breitenbuecher F, Wittke K, Du J, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114:2386–92.
Schnittger S, Bacher U, Haferlach C, Alpermann T, Kern W, Haferlach T. Diversity of the juxtamembrane and TKD1 mutations (exons 13–15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosomes Cancer. 2012;51:910–24.
Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, et al. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood. 2013; 121. doi:10.1182/blood-2012-07-442871.
Blau O, Berenstein R, Sindram A, Blau IW. Molecular analysis of different FLT3-ITD mutations in acute myeloid leukemia. Leuk Lymphoma. 2013;54:145–52.
Ozeki K, Kiyoi H, Hirose Y, Iwai M, Ninomiya M, Kodera Y, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 2004;103:1901–8.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–78.
Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3.
Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115:1425–32.
Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–83.
Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–76.
Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301.
DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–81.
DeAngelo D, Stone RM, Heaney ML, Nimer SD, Paquette R, Bruner-Klisovic R, et al. Phase II evaluation of the tyrosine kinase inhibitor MLN518 in patients with acute myeloid leukemia (AML) bearing a FLT3 internal tandem duplication (ITD) mutation. Blood (ASH Annual Meeting Abstracts). 2004; 104:Abstract 496.
Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. J Natl Cancer Inst. 2008;100:184–98.
Crump M, Hedley D, Kamel-Reid S, Leber B, Wells R, Brandwein J, et al. A randomized phase I clinical and biologic study of two schedules of sorafenib in patients with myelodysplastic syndrome or acute myeloid leukemia: a NCIC (National Cancer Institute of Canada) Clinical Trials Group Study. Leuk Lymphoma. 2010;51:252–60.
Pratz KW, Cortes J, Roboz GJ, Rao N, Arowojolu O, Stine A, et al. A pharmacodynamic study of the FLT3 inhibitor KW-2449 yields insight into the basis for clinical response. Blood. 2009;113:3938–46.
Metzelder S, Wang Y, Wollmer E, Wanzel M, Teichler S, Chaturvedi A, et al. Compassionate use of sorafenib in FLT3-ITD-positive acute myeloid leukemia: sustained regression before and after allogeneic stem cell transplantation. Blood. 2009;113:6567–71.
Sharma M, Ravandi F, Bayraktar UD, Chiattone A, Bashir Q, Giralt S, et al. Treatment of FLT3-ITD-positive acute myeloid leukemia relapsing after allogeneic stem cell transplantation with sorafenib. Biol Blood Marrow Transplant. 2011;17:1874–7.
Metzelder SK, Schroeder T, Finck A, Scholl S, Fey M, Götze K, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26:2353–9.
Kancha RK, Grundler R, Peschel C, Duyster J. Sensitivity toward sorafenib and sunitinib varies between different activating and drug-resistant FLT3-ITD mutations. Exp Hematol. 2007;35:1522–6.
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–605.
O’Farrell AM, Foran JM, Fiedler W, Serve H, Paquette RL, Cooper MA, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9:5465–76.
Fiedler W, Serve H, Döhner H, Schwittay M, Ottmann OG, O’Farrell AM, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–93.
Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54–60.
Fischer T, Stone RM, DeAngelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–45.
Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91.
Knapper S, Burnett AK, Littlewood T, Kell WJ, Agrawal S, Chopra R, et al. A phase 2 trial of the FLT3 inhibitor lestaurtinib (CEP701) as first-line treatment for older patients with acute myeloid leukemia not considered fit for intensive chemotherapy. Blood. 2006;108:3262–70.
Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114:2984–92.
Cortes JE, Perl AE, Dombret H, Kayser S, Steffen B, Rousselot P, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients ≥60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Blood (ASH Annual Meeting Abstracts). 2012; 120:Abstract 48.
Levis MJ, Perl AE, Dombret H, Döhner H, Steffen B, Rousselot P, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood (ASH Annual Meeting Abstracts). 2012; 120:Abstract 673.
Smith CC, Lasater E, Mccreery M, Lin K, Stewart W, Damon LE, et al. Crenolanib (CP-868,596) is a potent and selective type I FLT3 inhibitor that retains activity against AC220 resistance-causing FLT3 kinase domain mutants. Blood (ASH Annual Meeting Abstracts). 2012; 120:Abstract 141.
Lewis NL, Lewis LD, Eder JP, Reddy NJ, Guo F, Pierce KJ, et al. Phase I study of the safety, tolerability, and pharmacokinetics of oral CP-868,596, a highly specific platelet-derived growth factor receptor tyrosine kinase inhibitor in patients with advanced cancers. J Clin Oncol. 2009;27:5262–9.
Michael M, Vlahovic G, Khamly K, Pierce KJ, Guo F, Olszanski AJ. Phase Ib study of CP-868,596, a PDGFR inhibitor, combined with docetaxel with or without axitinib, a VEGFR inhibitor. Br J Cancer. 2010;103:1554–61.
Cortes J, Kantarjian H. How I treat newly diagnosed chronic phase CML. Blood. 2012;120:1390–7.
Ades L, Guerci A, Raffoux E, Sanz M, Chevallier P, Lapusan S, et al. Very long-term outcome of acute promyelocytic leukemia after treatment with all-trans retinoic acid and chemotherapy: the European APL Group experience. Blood. 2010;115:1690–6.
Ravandi F, Cortes JE, Jones D, Faderl S, Garcia-Manero G, Konopleva MY, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856–62.
Macdonald DA, Assouline SE, Brandwein J, Kamel-Reid S, Eisenhauer EA, Couban S, et al. A phase I/II study of sorafenib in combination with low dose cytarabine in elderly patients with acute myeloid leukemia or high-risk myelodysplastic syndrome from the National Cancer Institute of Canada Clinical Trials Group: trial IND.186. Leuk Lymphoma. 2013;54:760–6.
Serve H, Wagner R, Sauerland S, Brunnberg U, Krug U, Schaich M, et al. Sorafenib in combination with standard induction and consolidation therapy in elderly AML patients: results from a randomized, placebo-controlled phase II Trial. Blood (ASH Annual Meeting Abstracts). 2010; 116:Abstract 333.
Fiedler W, Kayser S, Kebenko M, Krauter J, Salih HR, Götze K, et al. Sunitinib and intensive chemotherapy in patients with acute myeloid leukemia and activating FLT3 mutations: results of the AMLSG 10-07 study. Blood (ASH Annual Meeting Abstracts). 2012; 120:Abstract 1483.
Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26:2061–8.
Kindler T, Lipka DB, Fischer T. FLT3 as a therapeutic target in AML: still challenging after all these years. Blood. 2010;116:5089–102.
Giles FJ, Cooper MA, Silverman L, Karp JE, Lancet JE, Zangari M, et al. Phase II study of SU5416—a small-molecule, vascular endothelial growth factor tyrosine-kinase receptor inhibitor—in patients with refractory myeloproliferative diseases. Cancer. 2003;97:1920–8.
Yin OQ, Wang Y, Schran H. A mechanism-based population pharmacokinetic model for characterizing time-dependent pharmacokinetics of midostaurin and its metabolites in human subjects. Clin Pharmacokinet. 2008;47:807–16.
Knapper S, Burnett AK, Hills R, Small D, Levis M. Lestaurtinib FLT3 inhibitory activity is modulated by concomitant azole therapy and may influence relapse risk. Blood (ASH Annual Meeting Abstracts). 2009; 114:Abstract 789.
Fischer T. Rethinking bioactivity of FLT3 inhibitors. Blood. 2011;117:3247–8.
Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–50.
Schaich M, Soucek S, Thiede C, Ehninger G, Illmer T. MDR1 and MRP1 gene expression are independent predictors for treatment outcome in adult acute myeloid leukaemia. Br J Haematol. 2005;128:324–32.
Chu SH, Small D. Mechanisms of resistance to FLT3 inhibitors. Drug Resist Updat. 2009;12:8–16.
Tai YT, Li XF, Breitkreutz I, Song W, Neri P, Catley L, et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006;66:6675–82.
Sexauer A, Perl A, Yang X, Borowitz M, Gocke C, Rajkhowa T, et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood. 2012;120:4205–14.
Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117:3286–93.
Grunwald MR, Levis MJ. Variations in FLT3 ligand levels during the course of AML treatment. J Clin Oncol (ASCO Annual Meeting Abstracts). 2013; 31:Abstract 7026.
Levis M. FLT3/ITD AML and the law of unintended consequences. Blood. 2011;117:6987–90.
Ravandi F, Alattar ML, Levis MJ, Garcia-Manero G, Richie MA, Daver NG, et al. Combination of sorafenib and 5-azacytidine has significant activity in patients with relapsed/refractory or untreated acute myeloid leukemia and FLT3-ITD mutation. Blood (ASH Annual Meeting Abstracts). 2012; 120:Abstract 1519.
Levis M, Tse KF, Smith BD, Garrett E, Small D. A FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood. 2001;98:885–7.
Clark JJ, Cools J, Curley DP, Yu JC, Lokker NA, Giese NA, et al. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood. 2004;104:2867–72.
Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003;102:646–51.
Cools J, Mentens N, Furet P, Fabbro D, Clark JJ, Griffin JD, et al. Prediction of resistance to small molecule FLT3 inhibitors: implications for molecularly targeted therapy of acute leukemia. Cancer Res. 2004;64:6385–9.
Heidel F, Solem FK, Breitenbuecher F, Lipka DB, Kasper S, Thiede MH, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107:293–300.
Albers C, Leischner H, Verbeek M, Yu C, Illert AL, Peschel C, et al. The secondary FLT3-ITD F691L mutation induces resistance to AC220 in FLT3-ITD(+) AML but retains in vitro sensitivity to PKC412 and Sunitinib. Leukemia. 2013; 27. doi:10.1038/leu.2013.14.
von Bubnoff N, Engh RA, Aberg E, Sanger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69:3032–41.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401.
Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367:2075–88.
Knapper S, Mills KI, Gilkes AF, Austin SJ, Walsh V, Burnett AK. The effects of lestaurtinib (CEP701) and PKC412 on primary AML blasts: the induction of cytotoxicity varies with dependence on FLT3 signaling in both FLT3-mutated and wild-type cases. Blood. 2006;108:3494–503.
Gu TL, Nardone J, Wang Y, Loriaux M, Villen J, Beausoleil S, et al. Survey of activated FLT3 signaling in leukemia. PLoS One. 2011;6:e19169.
Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–43.
Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Böhmer FD, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113:4063–73.
Schiller J, Praulich I, Krings Rocha C, Kreuzer KA. Patient-specific analysis of FLT3 internal tandem duplications for the prognostication and monitoring of acute myeloid leukemia. Eur J Haematol. 2012;89:53–62.
Parmar A, Marz S, Rushton S, Holzwarth C, Lind K, Kayser S, et al. Stromal niche cells protect early leukemic FLT3-ITD + progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res. 2011;71:4696–706.
Miething C, Grundler R, Mugler C, Brero S, Hoepfl J, Geigl J, et al. Retroviral insertional mutagenesis identifies RUNX genes involved in chronic myeloid leukemia disease persistence under imatinib treatment. Proc Natl Acad Sci. 2007;104:4594–9.
Donato NJ, Wu JY, Stapley J, Gallick G, Lin H. Arlinghaus, et al. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood. 2003;101:690–8.
Agarwal A, Eide CA, Harlow A, Corbin AS, Mauro MJ, Druker BJ, et al. An activating KRAS mutation in imatinib-resistant chronic myeloid leukemia. Leukemia. 2008;22:2269–72.
Nakano Y, Kiyoi H, Miyawaki S, Asou N, Ohno R, Saito H, et al. Molecular evolution of acute myeloid leukaemia in relapse: unstable N-ras and FLT3 genes compared with p53 gene. Br J Haematol. 1999;104:659–64.
Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109:1643–52.
Sargin B, Choudhary C, Crosetto N, Schmidt MH, Grundler R, Rensinghoff M, et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood. 2007;110:1004–12.
Zhang L, Ming L, Yu J. BH3 mimetics to improve cancer therapy; mechanisms and examples. Drug Resist Updat. 2007;10:207–17.
Kohl TM, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander SK, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–72.
Siendones E, Barbarroja N, Torres LA, Buendia P, Velasco F, Dorado G, et al. Inhibition of Flt3-activating mutations does not prevent constitutive activation of ERK/Akt/STAT pathways in some AML cells: a possible cause for the limited effectiveness of monotherapy with small-molecule inhibitors. Hematol Oncol. 2007;25:30–7.
Stölzel F, Steudel C, Oelschlägel U, Mohr B, Koch S, Ehninger G, et al. Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann Hematol. 2010;89:653–62.
Levis M, Small D. FLT3 tyrosine kinase inhibitors. Int J Hematol. 2005;82:100–7.
Whitman SP, Archer KJ, Feng L, Baldus C, Becknell B, Carlson BD, et al. Absence of the wild-type allele predicts poor prognosis in adult de novo acute myeloid leukemia with normal cytogenetics and the internal tandem duplication of FLT3: a cancer and leukemia group B study. Cancer Res. 2001;61:7233–9.
Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100:2393–8.
Shih LY, Huang CF, Wu JH, Lin TL, Dunn P, Wang PN, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100:2387–92.
Tiesmeier J, Müller-Tidow C, Westermann A, Czwalinna A, Hoffmann M, Krauter J, et al. Evolution of FLT3-ITD and D835 activating point mutations in relapsing acute myeloid leukemia and response to salvage therapy. Leuk Res. 2004;28:1069–74.
Gale RE, Green C, Allen C, Mead AJ, Burnett AK, Hills RK, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–84.
Nazha A, Cortes J, Faderl S, Pierce S, Daver N, Kadia T, et al. Activating internal tandem duplication mutations of the fms-like tyrosine kinase-3 (FLT3-ITD) at complete response and relapse in patients with acute myeloid leukemia. Haematologica. 2012;97:1242–5.
Conflict of interest
MJL serves as a consultant to Ambit Biosciences.
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
Grunwald, M.R., Levis, M.J. FLT3 inhibitors for acute myeloid leukemia: a review of their efficacy and mechanisms of resistance. Int J Hematol 97, 683–694 (2013). https://doi.org/10.1007/s12185-013-1334-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-013-1334-8