
Abstract
Geobacillus thermoglucosidasius Y4.1MC1 was isolated from a boiling spring in the lower geyser basin of Yellowstone National Park. This species is of interest because of its metabolic versatility. The genome consists of one circular chromosome of 3,840,330 bp and a circular plasmid of 71,617 bp with an average GC content of 44.01 %. The genome is available in the GenBank database (NC_014650.1 and NC_014651.1). In addition to the expected metabolic pathways for sugars and amino acids, the Y4.1MC1 genome codes for two separate carbon monoxide utilization pathways, an aerobic oxidation pathway and an anaerobic reductive acetyl CoA (Wood-Ljungdahl) pathway. This is the first report of a non-anaerobic organism with the Wood-Ljungdahl pathway. This anaerobic pathway permits the strain to utilize H2 and fix CO2 present in the hot spring environment. Y4.1MC1 and its related species may play a significant role in carbon capture and sequestration in thermophilic ecosystems and may open up new routes to produce biofuels and chemicals from CO, H2, and CO2.
Similar content being viewed by others
Introduction
Microbial consortia found in hot springs utilize carbon monoxide to obtain energy and fix carbon (reviewed in [1, 2]). The microbes of these consortia utilize the Wood-Ljungdahl pathway, a pathway distinct from the one utilized by the aerobic organisms that oxidize CO with molecular oxygen [3, 4]. The elucidation of the Wood-Ljungdahl pathway spans over 50 years of intensive research (reviewed in [5]. Researchers have isolated obligate anaerobic thermophiles from a number of environments including the first and most-studied acetogen, Moorella thermoacetica (formerly Clostridium thermocellum) whose genome was sequenced and analyzed [6]. Since then, other thermophilic CO oxidizers that utilize the Wood-Ljungdahl pathway have been isolated including Thermosinus carboxydivorans [7], Thermolithobacter carboxydivorans [8], and many from Yellowstone hot springs. All the isolated thermophiles with the Wood-Ljungdahl pathway are strict anaerobes, and no aerobic or facultative anaerobic thermophiles have been isolated. Here, we report the genome sequence of the first facultative anaerobe, a member of the genus Geobacillus, with the Wood-Ljungdahl pathway. In addition, this organism also possesses the aerobic CO oxidation pathway, a truly unusual occurrence.
The genus Geobacillus was established in 2001 to include aerobic and facultatively anaerobic, thermophilic spore-forming bacilli [9]. Geobacillus are obligately thermophilic (growth temperature range is 37–75 °C, with an optimum at 55–65 °C), and thus, most members are found in warm biotopes such as oil fields, compost heaps, geothermal areas, and most soil environments [10]. Surprisingly, Geobacillus are also found in cool biotopes, such as soil that never experiences elevated temperatures [11] or the bottom of the ocean. Geobacillus kaustophilus, which grows optimally at 60 °C with an upper limit of 74 °C, was isolated from the deepest sea mud of the Mariana Trench (~1 °C) [12]. As part of a project in conjunction with the Joint Genome Institute, Department of Energy, Lucigen Corp., isolated, characterized, and sequenced a number of new isolates from Yellowstone hot springs. The bacterial isolate Y4.1MC1 was one of four microorganisms isolated from Bath spring in Yellowstone National Park, Montana, USA, and submitted for whole genome sequencing. Geobacillus sp. Y4.1MC1 was collected from 88 °C water in the outflow channel of Bath hot spring in Yellowstone National Park and was classified as a Geobacillus sp. based on its isolation conditions and morphological similarity to other Yellowstone hot spring isolates such as Geobacillus species Y412MC61 (GenBank 544556), Y412MC52 (GenBank 550542), and Geobacillus thermoglucosidasius C56-YS93 (GenBank 634956). Sequencing and analysis of the Geobacillus sp. Y4.1MC1 genome identified it as the first sequenced CO oxidizer that is not a strict anaerobe.
Methods
Isolation, Growth Conditions, and DNA Isolation
G. thermoglucosidasius Y4.1MC1 (Y4.MC1) was isolated from a sample of hot spring water by enrichment and plating on YTP-2 medium at 70 °C [13] and maintained on tryptic soy broth without glucose (TSB) (Difco) agar plates. The culture is freely available from the Bacillus Genetic Stock Center (BGSC; www.bgsc.org). C5•6 Technologies Inc., Lucigen, the National Park Service, and the Joint Genome Institute have placed no restrictions on the use of the culture or sequence data. For preparation of genomic DNA, liter cultures of Y4.1MC1 were grown from a single colony in YTP-2 medium and collected by centrifugation. The cell concentrate was lysed using a combination of SDS and proteinase K, and the genomic DNA was isolated using a phenol/chloroform extraction. The genomic DNA was precipitated and treated with RNase to remove residual contaminating RNA.
Genome Sequencing and Assembly
The genome of Y4.1MC1 was sequenced at the Joint Genome Institute (JGI) using a combination of Illumina and 454 technologies. An Illumina GAii shotgun library with reads of 375 Mb, a 454 Titanium draft library with average read length of 510–525 bp bases, and a paired end 454 library with an average insert size of 18 kb were generated for this genome. All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov/. Illumina sequencing data was assembled with VELVET [14], and the consensus sequences were shredded into 1.5-kb overlapped fake reads and assembled together with the 454 data. Draft assemblies were based on 181.8 Mb 454 draft data and all of the 454 paired end data. Newbler parameters are Consed, 50–1350 g/ml [15]. The initial Newbler assembly contained 121 contigs in 18 scaffolds. We converted the initial 454 assembly into a phrap assembly by making fake reads from the consensus, collecting the read pairs in the 454 paired end library. The Phred/Phrap/Consed software package (www.phrap.com) was used for sequence assembly and quality assessment [16–18] in the following finishing process. Illumina data was used to correct potential base errors and increase consensus quality using the Polisher software developed at JGI (Alla Lapidus, unpublished). After the shotgun stage, reads were assembled with parallel Phrap (high-performance software; LLC). Possible mis-assemblies were corrected with Gap Resolution (Cliff Han, unpublished), Dupfinisher [19], or sequencing-cloned bridging PCR fragments with subcloning. Gaps between contigs were closed by editing in Consed, by PCR and by Bubble PCR primer walks. A total of 449 additional reactions and 9 shatter libraries were necessary to close gaps and to raise the quality of the finished sequence. The genome had an overall average error rate of 0.03 errors/10 kb.
Genome Annotation
Genes were identified using Prodigal [20] as part of the Oak Ridge National Laboratory genome annotation pipeline, followed by a round of manual curation using the JGI GenePRIMP pipeline [15]. The predicted CDSs were translated and used to search the National Center for Biotechnology Information (NCBI) nonredundant database, UniProt, TIGR-Fame, Pfam, PRIAM, KEGG, Cluster of Orthologous Groups (COG), and InterPro databases. These data sources were combined to assert a product description for each predicted protein. Noncoding genes and miscellaneous features were predicted using tRNAscan-SE [21], RNAMMer [22], Rfam [23], TMHMM [24], and SignalP [24].
The genome consists of one circular chromosome of 3,840,330 bp and a circular plasmid of 71,617 bp with an average GC content of 44.01 % (Table 1). The genome project is deposited in the Genomes OnLine Database (GOLD ID = Gc01645) [18, 25], and the complete genome sequence is deposited in GenBank. Of the 4031 genes predicted, 3910 were protein-coding genes and 121 RNAs; 241 pseudogenes were also identified (Table 2). The majority of the protein-coding genes (68.6 %) were assigned with a putative function, while the remaining ones were annotated as hypothetical proteins. The distribution of genes into COG functional categories is presented in Table 3.
Results
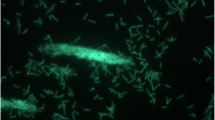
G. thermoglucosidasius Y4.1MC1 (Y4MC1) is one of a number of novel thermophilic species isolated from 88 °C water in the northern outflow channel of Bath hot spring (latitude 44.560318, longitude −110.8338344) in Yellowstone National Park under a sampling permit from the National Park Service. The temperature of Bath is 93 °C at the source, which is the boiling point at the prevailing elevation. The pH of the spring is 8.9 with SiO2 (244.8 mg/l) and Cl− (297.1 mg/l) as the dominant dissolved minerals (http://www.rcn.montana.edu/resources/features/feature.aspx?nav=11&id=4007). Y4.1MC1 is a gram-positive, rod-shaped facultative anaerobe with optimum growth temperature of 65 °C and maximum growth temperature of 75 °C. Y41MC1 appears to grow as a mixture of single cells and large clumps in liquid culture (Fig. 1).

Micrograph of Geobacillus thermoglucosidasius Y4.1MC1 cells showing individual cells and clumps of cells. Cells were grown in TSB plus 0.4 % glucose for 18 h at 70 °C. A 1.0-ml aliquot was removed, centrifuged, resuspended in 0.2 ml of sterile water, and stained using a 50 μM solution of SYTO® 9 fluorescent stain in sterile water (Molecular Probes). Dark field fluorescence microscopy was performed using a Nikon Eclipse TE2000-S epifluorescence microscope at × 2000 magnification using a high-pressure Hg light source and a 500-nm emission filter
A phylogenetic tree was constructed to identify the relationship of Y41MC1 to other members of the Geobacillus family (Fig. 2). The phylogeny of Y41MC1 was determined using its 16S ribosomal RNA (rRNA) gene sequence as well as those of the type strains of all validly described Geobacillus spp. The 16S rRNA gene sequences were aligned using MUSCLE [26], pairwise distances were estimated using the maximum composite likelihood (MCL) approach, and initial trees for heuristic search were obtained automatically by applying the neighbor-joining method in MEGA 5 [27]. The alignment and heuristic trees were then used to infer the phylogeny using the maximum likelihood method based on Tamura-Nei [28]. The phylogenetic tree identifies Y41MC1 as a G. thermoglucosidasius species. Average nucleotide identity (ANI) calculations [29] gave 99.1 % identity for the comparison of the Y41MC1 genome to the G. thermoglucosidasius C56-YS93 genome and an identical 99.1 % identity for the comparison of the Y41MC1 genome to the G. thermoglucosidasius TNO-09.020 genome. These values are above the proposed cutoff for separate species of 94 to 96 % [30] or the more recent proposed cutoff of 98.2 to 99.0 % [31].

Molecular phylogenetic analysis by maximum likelihood method was performed as detailed in text. The tree with the highest log likelihood (−4170.6736) is shown. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 18 nucleotide sequences. There were a total of 1570 positions in the final dataset. The type strains of all validly described species are included (NCBI accession numbers): G. caldoxylolyticus ATCC700356T (AF067651), Geobacillus galactosidasius CF1BT (AM408559), Geobacillus jurassicus DS1T (FN428697), G. kaustophilus NCIMB8547T (X60618), G. lituanicus N-3T (AY044055), Geobacillus stearothermophilus R-35646T (FN428694), G. subterraneus 34T (AF276306), Geobacillus thermantarcticus DSM9572T (FR749957), Geobacillus thermocatenulatus BGSC93A1T (AY608935), Geobacillus thermodenitrificans R-35647T (FN538993), G. thermoglucosidasius BGSC95A1T (FN428685), Geobacillus thermoleovorans DSM5366T (Z26923), Geobacillus toebii BK-1T (FN428690), Geobacillus uzenensis UT (AF276304), and Geobacillus vulcani 3S-1T (AJ293805). The 16S rRNA sequence of Paenibacillus lautus JCM9073T (AB073188) was used to root the tree
Insights from the Genome Sequence
Carbohydrate Metabolism
The secretome of Y4.1MC1 contains neither the xylanase nor arabinase found in other Geobacillus species [32, 33], suggesting a limited ability to utilize polysaccharide substrates. The organism does possess the ability to import and metabolize monosaccharides, oligosaccharides, sugar alcohols, and sugar acids. Many of these gene clusters are located within a ~120 kb region (2094175 through 2220362) that also contains clusters for urea utilization and peptide utilization.
Mannitol Metabolism
Y4.1MC1 possesses a gene cluster for a three-component phosphotransferase system (PTS) transporter systems that uses phosphoenolpyruvate to transport mannitol into the cell and phosphorylate it (GY4MC1_2238 and GY4MC1_2240), generating intracellular mannitol-1-phosphate. A MtlR family transcriptional regulator controls mannitol uptake (GY4MC1_2239). The mannitol utilization cluster also contains a gene coding for mannitol-1-phosphate 5-dehydrogenase, which converts the mannitol-1-phosphate to fructose-1-phosphate (GY4MC1_2237). Similar transport and metabolism clusters are used for fructose and cellobiose metabolism.
Gluconate Metabolism
Y4.1MC1 possesses an orthologous cluster for gluconate utilization (GY4MC1_2227 through GY4MC1_2229) similar to the GntU, GntK, and GntR cluster found in Escherichia coli [34]. Unlike the Bacillus subtilis gluconate utilization cluster [25], the Geobacillus cluster does not include a GntZ gene coding for 6-phosphogluconate dehydrogenase. The GntZ gene is present in another part of the genome (Y4MC1_1224).
Xylose Metabolism
In Y4.1MC1, a gene cluster codes for an aldose-1-epimerase (GY4MC1_2188), a three-component ABC transporter system for xylose (GY4MC1_2185 through GY4MC1_2187), a xylose isomerase (GY4MC1_2184), and a xylulose kinase (GY4MC1_2183). There are no genes for xylan degradation and utilization or arabinose utilization in the annotated Y4.1MC1 genome.
Cellobiose and Fructose Metabolism
In Y4.1MC1, gene clusters code for three-component PTS transporter systems that use phosphoenolpyruvate to transport the sugar into the cell and phosphorylate it, generating intracellular fructose-1-phosphate (GY4MC1_2122 through GY4MC1_2124) or cellobiose-6-phosphate (GY4MC1_2156 through GY4MC1_2158). A DeoR family transcriptional regulator controls fructose uptake (GY4MC1_2126). The six fructose utilization clusters also contain a gene coding for 1-phosphofructokinase (GY4MC1_2125), which converts fructose-1-phosphate to fructose-1,6-diphosphate. A GntR family transcriptional regulator controls cellobiose uptake (GY4MC1_2154). The cellobiose utilization clusters also contain a gene coding for 6-phospho-β-glucosidase (GY4MC1_2155), which converts cellobiose-6-phosphate to glucose and glucose-6-phosphate.
Inositol Phosphate Metabolism
The inositol phosphate utilization cluster (Table 4) has two separate parts under the control of a LacI family transcriptional regulator. Other Geobacillus species have been reported to possess similar clusters [35]. Y4.1MC1 does not possess any annotated phytase genes, but phytate may be converted to inositol phosphate by the secreted alkaline phosphatase (GY4MC1_2230).
Carbon Monoxide Metabolism Clusters
A unique feature of Y4.1MC1 is the presence of the Wood-Ljungdahl pathway, previously only found in strict anaerobes. A 15-kb cluster in Y4.1MC1 contains 15 genes coding for the anaerobic CO dehydrogenase/acetyl CoA synthase complex (DNA coordinates 1764093 to 1780695). This complex catalyzes the complex multistep anaerobic reactions that include oxidizing CO to CO2, formation of H2, and biosynthesis of acetyl CoA. BLASTn analysis shows that among Geobacillus species, only G. thermoglucosidasius strains possess this cluster. The DNA sequence of the Y4.1MC1 cluster is 98 % identical to the cluster present in G. thermoglucosidasius C56-YS93, isolated from Obsidian Hot Spring at Yellowstone National Park. Surprisingly, the next two closest matches were to two strict anaerobes, Thermoanaerobacter tengcongensis MB4 (now Caldanaerobacter subterraneus tengcongensis MB4T) with 70 % identity and 83 % coverage and M. thermoacetica ATCC 39073 with 73 % identity and 68 % coverage. Neighborhood analysis of orthologs shows that the organization of the Y4.1MC1 cluster is essentially identical to the M. thermoacetica ATCC 39073™ CO cluster (Table 5). The C. subterraneus tengcongensis CO cluster also shows the same organization as the Y4.1MC1 cluster (data not shown).
In addition to the anaerobic CO dehydrogenase/acetyl CoA synthase complex, Y4.1MC1 possesses genes coding for an aerobic-type carbon monoxide dehydrogenase complex GY4MC1_2422 through GY4MC1_2425 (Table 6). This cluster is found in two other G. thermoglucosidasius strains (NBRC 107763 and M10EXG). This complex allows oxidation of CO in the presence of oxygen or other electron acceptors such as nitrate or arsenate.
A carbonic anhydrase gene is located upstream of the CO dehydrogenase (GY4MC1_1804). Carbonic anhydrase allows efficient extraction of gaseous CO2 from the environment and conversion into the soluble carbonate anion. This particular carbonic anhydrase gene is present in all three sequenced G. thermoglucosidasius strains as well as Geobacillus caldoxylolyticus NBRC 107762. A structurally unrelated (11.9 % protein sequence identity) carbonic anhydrase is present in Geobacillus sp. C56-T3 and Geobacillus sp. JF8. The Y4.1MC1 carbonic anhydrase does not appear to be part of a carboxysome structure. Carboxysomes have an outer shell composed of protein subunits that contains carbonic anhydrase and RuBisCO [36]. A search of the genome of Y4.1MC1 reveals no ortholog of RuBisCO. The genome of Y4.1MC1 reveals the presence of two pairs of genes coding for microcompartment proteins (GY4MC1_1860–GY4MC1_1860 and GY4MC1_1866–GY4MC1_1867). The carbonic anhydrase gene is not part of either of these clusters. Analysis of the gene neighborhoods surrounding these microcompartments indicates that they are involved in the metabolism of 1,2-propanediol and ethylene glycol via a cobalamin-utilizing pathway. In place of the RuBisCO pathway, Y4.1MC1 possesses a malate synthase that converts acetyl CoA to malate (GY4MC1_1628) and a partial TCA cycle (malate dehydrogenase is not present in the genome) that allows conversion of malate into metabolites needed for production of amino acids, sugars, and other cellular components.
Conclusions
G. thermoglucosidasius Y4.1MC1 is a unique species, a facultative anaerobe capable of both aerobic and anaerobic oxidation of carbon monoxide. This is the first report of a facultative anaerobic thermophile possessing the Wood-Ljungdahl pathway. This anaerobic CO cluster not only imparts the ability to grow on CO but also confers the ability to grow on mixtures of H2 and CO2 [5, 6]. Because the hot springs of Yellowstone National Park produce primarily H2 and CO2 [37], Y4.1MC1 may utilize these two components in the otherwise nutrient-poor hot spring environment for both energy and cell mass production. Further work is needed to determine if Y4.1MC1 actively participates in the Bath microbial community or if the organism was deposited from another location via dust or spore dispersal. Our metagenomic analysis of the Bath hot spring community did not reveal Geobacillus signatures. Our experience with multiple Geobacillus strains has shown that lysis of cells and recovery of DNA is difficult from pure cultures grown in clear medium to a high cell density. These results suggest that the methods utilized for metagenomic DNA recovery may not be adequate for recovering intact DNA from Geobacillus cells present in the sample. Y4.1MC1 and related Geobacillus species may play a significant role in the capture and sequestration of CO2 generated in the hot springs. Additional work is needed to shed light on the ecological and physiological importance of these organisms. Y4.1MC1 offers the potential to produce new and novel biofuels and biopolymers from mixtures of CO, H2, and CO2. The ability of the organism to grow at temperatures approaching the boiling point makes it a perfect candidate for industrial processes. The ability of Y4.1MC1 to grow under aerobic conditions in liquid medium and on plates suggests that the metabolic engineering of Y4.1MC1 will be considerably easier than the engineering of strict anaerobes such as M. thermoacetica.
References
Techtmann S, Colman AS, Lebedinsky AV, Sokolova TG, Robb FT (2012) Evidence for horizontal gene transfer of anaerobic carbon monoxide dehydrogenases. Front Microbiol 3:132
Techtmann SM, Colman AS, Robb FT (2009) ‘That which does not kill us only makes us stronger’: the role of carbon monoxide in thermophilic microbial consortia. Environ Microbiol 11:1027–1037
King GM, Weber CF (2007) Distribution, diversity and ecology of aerobic CO-oxidizing bacteria. Nat Rev Microbiol 5:107–118
Kim YM, Park SW (2012) Microbiology and genetics of CO utilization in mycobacteria. Antonie Van Leeuwenhoek 101:685–700
Ragsdale SW, Pierce E (2008) Acetogenesis and the Wood-Ljungdahl pathway of CO(2) fixation. Biochim Biophys Acta 1784:1873–1898
Pierce E, Xie G, Barabote RD, Saunders E, Han CS et al (2008) The complete genome sequence of Moorella thermoacetica (f. Clostridium thermoaceticum). Environ Microbiol 10:2550–2573
Sokolova TG, Gonzalez JM, Kostrikina NA, Chernyh NA, Slepova TV et al (2004) Thermosinus carboxydivorans gen. nov., sp. nov., a new anaerobic, thermophilic, carbon-monoxide-oxidizing, hydrogenogenic bacterium from a hot pool of Yellowstone National Park. Int J Syst Evol Microbiol 54:2353–2359
Sokolova T, Hanel J, Onyenwoke RU, Reysenbach AL, Banta A et al (2007) Novel chemolithotrophic, thermophilic, anaerobic bacteria Thermolithobacter ferrireducens gen. nov., sp. nov. and Thermolithobacter carboxydivorans sp. nov. Extremophiles 11:145–157
Nazina TN, Tourova TP, Poltaraus AB, Novikova EV, Grigoryan AA et al (2001) Taxonomic study of aerobic thermophilic bacilli: descriptions of Geobacillus subterraneus gen. nov., sp. nov. and Geobacillus uzenensis sp. nov. from petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, G. th. Int J Syst Evol Microbiol 51:433–446
McMullan G, Christie JM, Rahman TJ, Banat IM, Ternan NG et al (2004) Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem Soc Trans 32:214–217
Rahman TJ, Marchant R, Banat IM (2004) Distribution and molecular investigation of highly thermophilic bacteria associated with cool soil environments. Biochem Soc Trans 32:209–213
Takami H, Nishi S, Lu J, Shimamura S, Takaki Y (2004) Genomic characterization of thermophilic Geobacillus species isolated from the deepest sea mud of the Mariana Trench. Extremophiles 8:351–356
Mead DA, Lucas S, Copeland A, Lapidus A, Cheng JF et al (2012) Complete genome sequence of Paenibacillus strain Y4.12MC10, a novel Paenibacillus lautus strain isolated from Obsidian hot spring in Yellowstone National Park. Stand Genomic Sci 6:381–400
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Pati A, Ivanova NN, Mikhailova N, Ovchinnikova G, Hooper SD et al (2010) GenePRIMP: a gene prediction improvement pipeline for prokaryotic genomes. Nat Methods 7:455–457
Ewing B, Hillier L, Wendl MC, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Han CS, Chain P (2006) Finishing repeat regions automatically with Dupfinisher. In: Arabnia HR, Valafar H (eds) Proceeding of the 2006 international conference on bioinformatics & computational biology. CSREA Press. pp. 141–146
Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW et al (2010) Prodigal prokaryotic dynamic programming genefinding algorithm. BMC Bioinforma 11:119
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T et al (2007) RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res 35:3100–3108
Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR (2003) Rfam: an RNA family database. Nucleic Acids Res 31:439–441
Krogh A, Larsson B, von Heijne G, Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580
Reizer A, Deutscher J, Saier MH Jr, Reizer J (1991) Analysis of the gluconate (gnt) operon of Bacillus subtilis. Mol Microbiol 5:1081–1089
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Tamura K, Peterson D, Peterson N, Stecher G, Nei M et al (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10:512–526
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P et al (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Richter M, Rosselló-Móra R (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci 106:19126–19131
Kim M, Oh HS, Park SC, Chun J (2014) Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int J Syst Evol Microbiol 64:346–351
Shulami S, Gat O, Sonenshein AL, Shoham Y (1999) The glucuronic acid utilization gene cluster from Bacillus stearothermophilus T-6. J Bacteriol 181:3695–3704
Shulami S, Raz-Pasteur A, Tabachnikov O, Gilead-Gropper S, Shner I et al (2011) The L-Arabinan utilization system of Geobacillus stearothermophilus. J Bacteriol 193:2838–2850
Tong S, Porco A, Isturiz T, Conway T (1996) Cloning and molecular genetic characterization of the Escherichia coli gntR, gntK, and gntU genes of GntI, the main system for gluconate metabolism. J Bacteriol 178:3260–3269
Yoshida K, Sanbongi A, Murakami A, Suzuki H, Takenaka S et al (2012) Three inositol dehydrogenases involved in utilization and interconversion of inositol stereoisomers in a thermophile, Geobacillus kaustophilus HTA426. Microbiology 158:1942–1952
Yeates TO, Kerfeld CA, Heinhorst S, Cannon GC, Shively JM (2008) Protein-based organelles in bacteria: carboxysomes and related microcompartments. Nat Rev Microbiol 6:681–691
Spear JR, Walker JJ, McCollom TM, Pace NR (2005) Hydrogen and bioenergetics in the Yellowstone geothermal ecosystem. Proc Natl Acad Sci U S A 102:2555–2560
Acknowledgments
This work was funded by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494). Sequencing work was performed under the auspices of the US Department of Energy’s Office of Science, Biological and Environmental Research Program, and by the University of California, Lawrence Berkeley National Laboratory under contract No. DE-AC02-05CH11231, Lawrence Livermore National Laboratory under Contract No. DE-AC52-07NA27344, and Los Alamos National Laboratory under contract No. DE-AC02-06NA25396.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Brumm, P., Land, M.L., Hauser, L.J. et al. Complete Genome Sequence of Geobacillus strain Y4.1MC1, a Novel CO-Utilizing Geobacillus thermoglucosidasius Strain Isolated from Bath Hot Spring in Yellowstone National Park. Bioenerg. Res. 8, 1039–1045 (2015). https://doi.org/10.1007/s12155-015-9585-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12155-015-9585-2