
Abstract
Correlative light and electron microscopy exploits the advantages of optical methods, such as multicolour probes and their use in hydrated live biological samples, to locate functional units, which are then correlated with structural details that can be revealed by the superior resolution of electron microscopes. One difficulty is locating the area imaged by the electron beam in the much larger optical field of view. Multifunctional probes that can be imaged in both modalities and thus register the two images are required. Phosphor materials give cathodoluminescence (CL) optical emissions under electron excitation. Lanthanum phosphate containing thulium or terbium or europium emits narrow bands in the blue, green and red regions of the CL spectrum; they may be synthesised with very uniform-sized crystals in the 10- to 50-nm range. Such crystals can be imaged by CL in the electron microscope, at resolutions limited by the particle size, and with colour discrimination to identify different probes. These materials also give emissions in the optical microscope, by multiphoton excitation. They have been deposited on the surface of glioblastoma cells and imaged by CL. Gadolinium oxysulphide doped with terbium emits green photons by either ultraviolet or electron excitation. Sixty-nanometre crystals of this phosphor have been imaged in the atmospheric scanning electron microscope (JEOL ClairScope). This probe and microscope combination allow correlative imaging in hydrated samples. Phosphor probes should prove to be very useful in correlative light and electron microscopy, as fiducial markers to assist in image registration, and in high/super-resolution imaging studies.
Similar content being viewed by others


Introduction
Most antibody localisation imaging techniques in biology have been based on optical microscopy, where the Rayleigh criterion
relates resolution (d) to the wavelength (λ) and numerical aperture (NA) of the objective lens. In practice, using high power microscope objectives, 200-nm resolution can be achieved using standard fluorescence or confocal microscopy. For this purpose, a wide range of fluorescent probes having varied colours and functions have been developed [12]. More advanced super-resolution microscopic (SRM) methods can extend the limits to a few tens of nanometres [16], but generally require more specialised equipment and probes.
The resolution of electron microscopes (EMs) is limited by the instrumental design and aberrations and is of the order of 1 nm for biological materials, depending on the sample nature and any staining methodology. It may be employed for high-resolution imaging but suffers from the lack of multifunctional probes. In particular, using this high resolution for colocalisation studies of different probes is difficult in the monochrome images obtained and is typically limited to thin sections or surfaces of heavily processed samples.
An alternative methodology that combines the resolution of electron microscopes with the wavelength discrimination of optical detection is cathodoluminescence (CL) [13]. This employs phosphor particles as probes and detects the photons emitted under electron bombardment. The emission wavelength depends on the phosphor dopant, which is generally a lanthanide element in a chemical matrix; for example, europium has bands mostly in the red, terbium in the green and thulium in the blue. The emission bands are narrow and can be tuned to some extent by choice of the matrix structure. The detection system can employ standard optical filters to distinguish different phosphors; the detector is synchronised with the electron beam scan, and thus, the resolution of the CL image is limited by the phosphor particle size. This high-resolution imaging modality can be thought of as electron-generated super resolution microscopy or eSRM.
Phosphors are very widely used in display technology, and a vast range of such materials has been synthesised in sizes down to a few nanometres [7]. Many can also be used in optical microscopy. Their properties can be adjusted by varying the matrix, the dopant and sensitiser elements [24]. In some cases, the particles can be detected by conventional single photon excitation, for example by ultraviolet excitation and detection in the visible (“downconversion”; [14]), but more often, multiphoton excitation (“upconversion”) in a confocal microscope is required. For biological applications, the phosphor is required to be as small as possible, stable in the electron beam and biologically inert; they must be capable of being conjugated to bioprobe molecules such as antibodies, aptamers and nanobodies. Many protocols are available that have been developed for use with particles such as quantum dots [9] that rely on silanisation to coat the particle surface with, e.g. aminopropyl triethoxysilane as shown on the left, then conjugation of the bioprobe with a carboxyl-to-amine cross-linking agent such as ethyl (dimethylaminopropyl) carbodiimide

Another application where the bifunctional nature of phosphor probes can be useful is in correlative light and electron microscopy (CLEM) [1, 27, 4, 11] where ultrastructural information is obtained by EM after optical microscopy has obtained the lower resolution data required for full description of the biological sample. One difficulty in CLEM is that the sample processing techniques can damage and distort the sample so that correlation of the two image modalities is difficult. This problem has been overcome to some extent by care in the sample processing step [23], but the use of fiducial markers that can be identified in both image types is extremely helpful. The nanophosphors can be used nonspecifically, by applying the particles from suspension before the drying process, or specifically by labelling with the conjugated bioprobe.
There have been a few reports of the use of nanometre-sized phosphors for imaging. Quantum dots (QDs) have been employed for correlative fluorescence and transmission electron microscopy of labelled nuclei in thin sections [20], but here, TEM detection was just by conventional electron contrast. Cathodoluminescence has been detected from QDs [5], but these II–VI semiconductors have relatively poor stability and spatial resolution under electron excitation ([17] and citations therein). Two types of nanodiamonds with cerium-doped lutetium aluminium garnet nanoparticles have been shown to give red, blue and green emissions from 40- to 80-nm crystals with good stability under the electron beam [8].
The requirements for cathodoluminescent phosphors differ somewhat from those needed for conventional fluorescence probes; the size and shape of the nanoparticles are as important as the chemical composition, of the matrix and of the dopants. We have found that commercial phosphor samples optimised for light microscopy are too weakly emitting in the electron beam and have high shape factors. We have chosen to synthesise lanthanum phosphate nanoparticles for these CLEM studies, using the technique described in the reference [26]. This generates the desired monoclinic monazite structure of LaPO4, and the size can be varied by adjusting the hydrothermal conditions and the composition of the reactants employed. These materials are colourless, have absorption bands in the vacuum ultraviolet [25] and can be upconverted by infrared illumination to give visible emissions.
The characteristics of these probes have been investigated by conventional CL microscopy using a field emission scanning electron microscope (FEGSEM). Their size and optical properties have been examined on silicon surfaces, and on fixed, dried cell membranes. The lanthanide elements provide good contrast by secondary electron detection, as well as the specificity of the optical emissions so that different phosphors can be identified.
The requirement that biological materials must be fixed and fully dehydrated for examination in the SEM vacuum chamber can lead to sample distortions. This can be avoided by the use of an atmospheric scanning electron microscope (ASEM) [19, 18]. Hydrated samples are held outside the EM vacuum chamber in a liquid-filled dish with an electron-permeable silicon nitride window. Optical imaging can be performed with the fluorescence microscope mounted above the dish, using a water dipping lens, before switching to the inverted SEM mode for correlative imaging of areas of interest in the sample on the silicon nitride film.
Methods
Cell culture for CL and SEM
U251 glioblastoma cells were seeded at a low density on to 12-mm diameter glass cover slips precoated with polylysine (P8920, Sigma-Aldrich) and cultured in RPMI medium (Gibco) for 48 h. They were then fixed in 4 % paraformaldehyde and dried by immersion in successively higher concentrations of ethanol and finally in hexamethyldisilazine (Sigma-Aldrich). The glass was attached to a pin stub with silver Dag (Agar Scientific Ltd.).
Phosphor sample preparation for light and electron microscopy
Phosphor probes were synthesised by a method developed to generate uniform small particles [26]. Briefly, lanthanum phosphate was precipitated in the presence of the respective dopants on quartz wool, fired at temperatures up to 1050 °C and removed from the substrate by sonication in isopropanol.
A suspension of nanoparticles in ethanol was freshly probe sonicated for 5 min, and then, 10 μl was applied to a silicon slab fixed to a pin stub using silver Dag and dried at 50 °C. For light microscopy, the suspension was applied to polylysine-coated glass cover slips (Zeiss #1.5) and mounted on a slide with ~50 μm water or buffer in between the glass surfaces.
Multiphoton confocal imaging
Multiphoton images were obtained using a Zeiss LSM780 confocal microscope, using a ×63 PlanApochromat water immersion (NA 1.2) objective, equipped with a Coherent Chameleon Ultra Multi-photon (MP) laser in addition to the conventional visible lasers. Emissions were detected by nondescanned detectors [3], the emitted light being split between blue (<485 nm), green (500–620 nm) and red (>620 nm) channels for simultaneous recording. A detector was also available for transmitted light observations. The laser could be tuned between 690 and 1040 nm; the laser intensity was adjusted to show signals in the detectors without obvious perturbation to the sample, seen as distortions in the transmission image.
Cathodoluminescence spectroscopy and imaging of nanoparticles
The CL microscope is constructed around a 7600 F electron microscope (JEOL (UK) Ltd), equipped with an Alto low-temperature stage and MonoCL4 system (Gatan UK Ltd.). The MonoCL4 detector is a high sensitivity photomultiplier, mounted on the exit from a grating monochromator with 300 or 1200 lines per cm. It was Peltier cooled to −30 °C and was usually operated at 1400 V. Light was collected by a paraboloid mirror that could be inserted above the sample, the electron beam passing through a 1.3-mm port above the specimen. The photons are reflected down a lightpipe, either via a monochromator for spectrum scanning or directly into the detector housing where the wavelength may be selected by bandpass filters (UV, 360WB50; green 535BP45; far-red 700BP50; all from Omega Filters, Brattleboro VT, USA). SEM signals from the secondary electron detector, and CL pulse mode output, were recorded by Digital Microscope software that controlled the scanning of the 7600 F. Images were typically obtained at ×75,000 magnification, using 256 × 256-pixel images giving 5.56 nm per pixel; pixel dwell times were of the order of 1~4 ms.
Atmospheric scanning electron microscopy
Images were obtained using a JEOL JASM-6200 instrument, modified by replacing the original colour camera by a charge-coupled device (Hamamatsu ORCA-ER). Sets of overlapping exposures were taken to observe the entire dish window, using times of a few milliseconds for reflectance and fluorescence; illumination was via an optical fibre from the Olympus mercury lamp. Fluorescence and CL imaging were performed with a DAPI filter set (emission 420-nm-long pass). CL imaging was performed by selecting a suitable electron microscope magnification (×1200 or ×5000) and stage position, setting a long camera exposure time, and allowing the ASEM to perform a single scan with the electron detector disabled.
Biological samples to be used in the ASEM were prepared in ClairScope dishes with the same protocols as used for multiphoton imaging. After fixation, the liquid was replaced by deionised water, and the phosphor materials were then applied from aqueous suspension after probe sonication. The excess phosphor was carefully removed by pipette and the dish inserted in the microscope for examination using a ×40 NA0.80 water dipping lens.
Image analysis
Pulse mode CL images from the MonoCL4 system and multiphoton images were analysed by a version of single-particle fluorescence imaging (SPFI) [2]. Briefly, values of pixels around the approximate centroid of a particle are fitted by least-squares methods to a 2D Gaussian:
where C(i,j) are the counts in the pixel at (row i, column j), B n is the background level around particle n, A n is the amplitude of the Gaussian (the intensity of the CL signal), {X n , Y n } are the coordinates of the centroid and W n is the width of the Gaussian which is assumed to be symmetrical, e.g. the CL originates from a point source. The variable parameters are B, A, X, Y and W; the weights of the data points Z(i,j) are taken to be √Z(i,j) as defined by Poissonian statistics. The background term B n can be replaced by a sloping background having adjustable direction and gradient; this is appropriate for CL images, as seen in Fig. 2.
Results
Phosphor probe spectroscopy and imaging
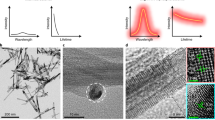
Figure 1 shows examples of the smallest nanophosphors studied, having diameters 15–25 nm. Spectra taken from large aggregates of LaPO4/Eu, Tb and Tm show the major peaks at 620, 545 and 350 nm, respectively. Inset in these spectra is small areas of the image pairs taken with 2-ms dwell time per pixel, left hand SEM and right hand CL in false colour. Most of the objects are small aggregates, and at 5 kV very small single particles are not detected. Lower energy (3 kV) electrons can allow CL detection, but focus becomes more difficult at low voltage and increased working distance (10 mm).

Cathodoluminescence emission spectra of LaPO4 nanoparticles on silicon stubs; the dopants are (top) Eu, (middle) Tb, (bottom) Tm. Inset are SEM and false-colour CL images, and histograms of the particle sizes, measured from SEM images of ~50 particles. The smallest crystals (~15 nm, for example green image, top left particle) are not detected under the conditions used (5 kV, 2 ms pixel dwell time)
Cell imaging
Initial tests were performed using silica-coated but non-derivatised phosphor particles on fixed cells. This was done so that the density of particles could be varied without performing biological modifications such as upregulation, and the aggregation state of the bound particles was not affected by any cell receptor oligomerisation. Also, large flat cells could be employed; U251 glioblastoma cells were grown on glass coverslips, fixed and dehydrated; then, a suspension of the chosen probe in ethanol was applied. Typical images are seen in Fig. 2, using the JEOL 7600 F FEGSEM at 5 kV and ×2000 magnification, with the LEI secondary electron detector. The box shows the area that is imaged at ×33,000 magnification in b for SEM and c for CL, in false-colour as shown in the colour panel. A bright background is seen near the nucleus of a post-mitotic cell at lower right, probably due to secondary electron effects in the sloping part of the membrane. Nevertheless, the blue-emitting nanophosphor can be easily distinguished.

Images of phosphors: a U251 cells grown on polylysine coated glass, with non-derivatised phosphor probes deposited at low density, dried and gold-palladium coated (2 nm thick). b SEM image at 5 kV and ×33,000 magnification, showing the edge of the nucleus of a post-mitotic cell, and c the false-colour image of CL emission with colour table in the inset. d Red- and blue-emitting particles deposited on silicon, and sequentially imaged with red, green and blue CL filter sets; the two phosphors can be readily distinguished. e Green- (arrowed) and blue-emitting particles imaged by the same CL filter sets, can be distinguished by their green/blue ratios
Imaging of mixed phosphors is accomplished by taking separate images with red, blue and green bandpass filters in the lightpath from the mirror to the PMT, and then combining the images with offsets to allow for the drift in the position. The offsets are found by comparing the positions of the particles in the three images, as determined by SPFI analysis. Addition of three images results in lower signal to noise, but red and blue phosphors can be readily distinguished as there is little overlap between the spectra as seen in Fig. 1. The green and blue phosphors have overlapping peaks in the 470-nm region and thus are much more difficult to identify; this can only be done by determining the green/blue intensity ratios, akin to spectral unmixing. Those showing blue have blue/green signal ratios greater than 2.5, while the arrowheads mark particles having signal ratios 1.5 ± 0.5 which can be assigned as green phosphors. The mean size of these particles estimated using SPFI is 37 ± 13 nm.
Multiphoton imaging
Images were obtained for the LaPO4 phosphor particles, using the polylysine-coated slides described in “Methods”. Laser power was adjusted to show non-saturating signals in the detectors when using 740-nm excitation (Fig. 3a), and then, an excitation fingerprint was obtained from 700 to 860 nm, as shown in Fig. 3b. The images were quantified by SPFI methods; the total signal under each 2D-Gaussian peak is normalised across the wavelength range, corrected for the laser power variation, and the resulting intensity fraction at each wavelength is plotted as a mean with standard deviation for 10 particles having medium intensity values. These were used to ensure adequate signal-to-noise ratios at the longer laser wavelengths; the same trends were observed for smaller particles (data not shown). The maximum signal is obtained at 720 nm, and good intensity can be observed across a wide range for these phosphors. It is noticeable that the response drops off much more rapidly for the blue lines as shown by the blue squares. A red analysis was not performed as the multiphoton system blocks emissions above 690 nm.

Multiphoton confocal image (top left) of LaPO4/Tb phosphors on a glass coverslip, using 740-nm excitation and a non-descanned detector collecting green light. Varying the excitation wavelength between 700 and 860 nm gives the profile shown (top right, green circles; mean of 10 particles of medium intensity), while detecting the emission using blue filters gives the profile shown as blue squares. Lower left the change of intensity shown by three single particles of low (pale), medium (green) and high (dark) intensity as the laser power is increased; at high power, the emission becomes saturated, while at low power, confocal image noise dominates. The dashed lines are linear fits to the log-log plots; the mean gradient is 1.9 suggesting a two-photon process. Lower right the emission spectra recorded by Zeiss spectral detector, mean of 8 particles of medium intensity. Dashed lines indicate the positions of peaks in the CL spectrum (Fig. 1). The peaks from these samples are relatively broad, compared with those found for other phosphor, e.g. Gd2O2S/Pr as shown in the inset
The power dependence of the emission from the particles was determined by ramping the laser intensity from 0.5 to 4 %; higher intensities caused saturation of the signals (Fig. 3c), possibly caused by heat damage at the higher power. Again, the signal was quantified by SPFI; at low power, the signals were dominated by noise, but log-log plots of signal against % power showed straight line behaviour, and the gradient reflects the number of infrared photons per multiphoton event [22]. The average for 60 particles was 1.90~0.28, suggesting a two-photon process.
The emission spectrum of the phosphors can be observed using a spectral detector, where the emission wavelengths are spread over 32 segments of a linear detector (typically 8 nm per segment). As the efficiency of this detector channel is lower than that of the NDD detectors employed for the excitation and power dependence measurements, higher laser powers were used to offset the lower signal-to-noise ratio obtained in individual segments of the detector. The emission spectra are shown in Fig. 3d, averaged for 8 particles that showed similar total intensity. The peaks correspond to those found in the CL emission spectrum (Fig. 1) but are broader, and the intensities vary between particles. An example of a different phosphor (Gd2O2S/Pr) having larger crystals (>50 nm) is shown in inset, and here, the peaks are much narrower and more intense.
Correlative ASEM imaging
U251 cells were grown on polylysine-coated ASEM dishes at low density, fixed and then phosphor particles in aqueous suspension were allowed to settle on the surface. In this case, somewhat larger particles of a terbium-doped gadolinium oxysulphide phosphor were used (Gd2O2S/Tb, mean diameter 60 nm, but with larger crystals present up to 1 μm). This material was used because it has a fluorescence emission under UV excitation.
The window was imaged first in reflectance mode to identify suitable areas for SEM and CL imaging (Fig. 4a). Figure 4b displays fluorescence (in blue false colour as shown in the inset) from UV excitation and long pass blue emission (‘DAPI’ type filter set with a 420-nm LP emission filter). This area is the box drawn in 4a and shows the high background at the top where there is autofluorescence from the cells, and a lack of fluorescence from the phosphor particles on top of the cells where they are not in focus. When imaged under CL conditions (20 kV), the image (c) is obtained, in blue-green-yellow-white false colour (inset).

Images of U251 cells with gadolinium oxysulfide nanoparticles, recorded by ASEM. a a reflectance image showing the glioblastoma cells growing in layers at the top of the image. The line down the right hand side is the edge of the silicon nitride window. b The fluorescence image of the box in (a), using UV excitation and a DAPI (420-nm-long pass) filter set. c The same area using 20-kV electron excitation and CL emission detection. Both panels are in false-colour as indicated by the inset colour bars
Two aspects of these images should be noted. The CL image shows higher sensitivity as many more particles are visible: this is partly due to the lower background signal, which is more apparent on the cell due to autofluorescence. Second, the depth of focus in CL mode is much greater: small particles on the lamellipodium near the top of the image, which are in a different Z-plane and hence are invisible by fluorescence, can be seen by CL as the electron beam has a much greater depth of focus.
Discussion
Correlative imaging, using the high resolution of the electron microscope in tandem with the flexibility of fluorescence methods, is a powerful technique that requires careful matching of the images from the two modalities. The images from optical microscopes with high-quality corrected objective lenses are only very slightly imperfect due to, e.g. chromatic aberration, but electron microscope images suffer from distortions due to charging and the local sample geometry. In order to match the two results, it is helpful to have bifunctional “fiducial” markers that are visible in both the optical and electronic images [21].
Although research in phosphor particles has been intense over the last 20 years, the use of nanophosphors in biological imaging is a new field [6]. Using the scanning method, XY resolution is limited only by particle size, provided the signal to noise is high enough. Using conventional models for the interaction between the electron beam and the solid particle, it can be predicted that greater electron interaction and hence higher CL signals will be obtained at low voltages (<5 kV). However, the greater interaction volume leads to greater damage and hence fading of the CL signal. With the type of phosphors used here, it would seem that 5 kV offers the best balance between CL signal and beam damage, and that 30 nm is a suitable particle size for positive detection with current CL equipment. The effects of sensitiser and dopant concentrations on the size detection limit have not been fully explored. Here, it is clearly demonstrated that we can see individual CL probes that can act as fiducial markers at high precision. Naturally, the probes can also be used to identify cells of interest if doped with these probes, e.g. by microinjection or phagositosis, at which point, the concentration of probes could be much greater and thus be imaged with a wide range of varying conditions including voltages.
This particle size is too large to identify all targets in crowded areas of the cell membrane, when using specific labels. Ideally, the nanophosphor particles should be as small as possible. A Fab fragment would be ~5-nm diameter; so, a nanophosphor particle should be smaller than this to prevent steric hindrance and maximise the binding potential and achievable resolution. The current probes will be suitable for imaging at the best resolution obtained by existing SRM methods, and thus, the resolution will be limited by the probe size. In the case of the ASEM, the best resolution using gold nanoparticles in an aqueous environment is about 15 nm.
The phosphors used here were often seen as small aggregates (Fig. 1), resulting from the high temperature synthesis step. These are adequate for use as fiducial markers; however, employment as specific probes will require single crystals of uniform size; they should also be of approximately spherical shape, as CL intensity depends on the depth of interaction with the electron beam, and asymmetric particles give different signals depending on orientation. Single particles can be separated by, e.g. centrifugation, but this is wasteful of material; modified high temperature processing should improve this. Very small monodisperse particles of lanthanum phosphate (6 nm) have been synthesised at lower temperatures [25]; however, these have not yet been shown to be useful for CL imaging. The gadolinium oxysulphide used for ASEM imaging was quite inhomogeneous, containing some large (1 μm) crystals and aggregates as can be seen in Fig. 4. This material was not optimised for use in bioimaging, and we are investigating the use of yttrium oxysulphide instead [10].
The ability to distinguish the different coloured probes is an advantage for the use of nanophosphor probes when compared with conventional heavy metal or immunogold staining. It is possible to distinguish different targets by using different nanogold probes, e.g. 10 and 20 nm. CL probes can have the same size so there will be no steric differentiation, are less dependent on perfect alignment of the electron microscope to distinguish different probes, and several different coloured phosphors could be employed simultaneously. When compared with quantum dots, phosphor colour depends on the chemistry of the material used and not on the size of the particle.
The CL equipment must be further developed before multicolour probe imaging can be performed routinely; in particular, the single PMT detector used here necessitates separate scans to be taken with each emission filter. Thus, sample drift needs to be corrected for between frames, resulting in slight loss of registration and weaker signal to noise. The simple addition of two PMT detectors with the appropriate optical splitter would significantly improve imaging conditions, and thus minimise effects of sample drift and electron beam damage which can cause mistakes in probe identification. The CL optical filters can be chosen to pass the primary peak of the emission spectrum and block the primary and secondary peaks of the other phosphors, but at some cost of efficiency. However, it is expected that the signal to noise gain would outweigh any loss from satellite emission bands.
An important aspect for future applications comes from the ability to image the probes using light microscopy. Here, we have demonstrated the ability to excite the probes using a standard multi-photon microscope. The probes were highly emitting upon excitation at 740 nm and could be readily identified using typical laser settings, which give a pixel dwell time of around 1–2 μs and are compatible with live cell imaging. However, some lanthanide probes had markedly longer lifetimes that resulted in poorer image quality. The europium and thulium doped probes were well suited for light microscopy as they demonstrated relatively fast lifetimes that gave rise to punctate images with no ‘comet’ effects, and thus behaved like standard organic fluorescent dyes under these conditions. However, some terbium phosphors had significantly longer lifetimes as could be seen from the “comet” trail left post-excitation when using non-descanned detectors. Although this is not ideal for imaging biological samples, the long decay tails found in the images did allow the approximate lifetimes of these probes to be calculated; the trails can also act as fingerprints to identify unambiguously the emission from the phosphors in cells that show artefacts from, e.g. lysosomal storage bodies.
The JEOL ClairScope ASEM is a novel correlative microscope that allows imaging of hydrated samples in optical and SEM modes. Fluorescent staining can permit identification of a suitable area for study prior to staining for SEM, but the heavy metal protocols will often destroy simple fluorescence. Introduction of stable phosphor nanoparticles, by antibody or by phagocytosis, can assist in identification of the target area. It is apparent from Fig. 4 that a greater depth of focus is achieved with CL imaging, so the use of phosphor particles should allow a greater degree of 3D imaging than can be achieved with pure SEM. The ASEM can image to a maximum depth of ~3 μm using 30 kV electrons, but this limit is set by the two-way passage of electrons through the aqueous phase. In CL imaging, the electrons will only have one journey, so greater depth into the sample may be achieved at some cost of resolution. It is to be hoped that electron excited super resolution microscopy (eSRM) will achieve resolutions covering the gap between pure optical and pure electron imaging, in both vacuum and hydrated environments.
This bifunctionality promises to make nanophosphors an ideal fiducial marker for CLEM. While the need for multiphoton light microscopy is a relatively expensive option for fluorescence imaging, further developments of phosphor probes will increase their suitability and sensitivity for standard light microscopy [7]. New combinations of electron and optical microscopes are being introduced [15], but in all cases, there is a clear need for fiducial markers. The photo- and electron stability of the probes compared to standard organic dyes also makes these highly desirable for future studies and development.
References
Agronskaia AV, Valentijn JA, van Driel LF, Schneijdenberg CTWM, Humbel BM, van Bergen en Henegouwen PMP, Verkleij AJ, Koster AJ, Gerritsen HC (2008) Integrated fluorescence and transmission electron microscopy. J Struct Biol 164:183–189
Anderson CM, Georgiou GN, Morrison IEG, Stevenson GVW, Cherry RJ (1992) Tracking of cell surface receptors by fluorescence digital imaging microscopy using a charge-coupled device camera. Low density lipoprotein and influenza virus mobility at 4C. J Cell Sci 101:415–425
Benninger RKP, Piston DW (2013) Two-photon excitation microscopy for the study of living cells and tissues. Curr Protoc Cell Biol 59:4.11.1–24
Bos E, SantAnna C, Gnaegi H, Pinto RF, Ravelli RB, Koster AJ, de Souza W, Peters PJ (2011) A new approach to improve the quality of ultrathin cryosections; its use for immunogold EM and correlative electron cryotomography. J Struct Biol 175:62–72
Deerinck TJ, Giepmans BN, Smarr BL, Martone ME, Ellisman MH (2007) Light and electron microscopic localisation of multiple proteins using quantum dots. Methods Mol Biol 374:43–53
Furukawa T, Niioka H, Ichimiya M, Nagata T, Ashida M, Araki T, Hashimoto M (2013) High resolution microscopy for biological specimens via cathodoluminescence of Eu- and Zn-doped Y2O3 nanophosphors. Opt Express 21:25655–25663
Gargas DJ, Chan EM, Ostrowski AD, Aloni S, Altoe VP, Barnard ES, Sanii B, Urban JJ, Milliron DJ, Cohen BE, Shuck PJ (2013) Engineering bright sub-10nm upconverting nanocrystals for single molecule imaging. Nat Nanotechnol 1:300–305
Glenn DR, Zhang H, Kasthuri N, Schalek R, Lo PK, Trifonov AS, Park H, Lichtman JW, Walsworth RL (2012) Correlative light and electron microscopy using cathodoluminescence from nanoparticles with distinguishable colours. Sci Rep 2(865):1–5
Hermanson G (2013) Bioconjugate Techniques, Third Edition. pub. Academic Press 535–587.
Hirai T, Orikoshi T (2004) Preparation of yttrium oxysulfide phosphor nanoparticles with infrared-to-green and –blue upconversion emission using an emulsion liquid membrane system. J Colloid Interface Sci 273:470–477
Hodgson L, Tavare J, Verkade P (2014) Development of a quantitative correlative light electron microscopy technique to study GLUT4 trafficking. Protoplasma 251:403–416
Johnson I (1998) Fluorescent probes for living cells. Histochem J 30(3):123–140
Kimura E, Sekiguchi T, Oikawa H, Niitsuma J, Nakayama Y, Suzuki H, Kimura M, Fujii K, Ushiki T (2004) Cathodoluminescence imaging for identifying uptaken fluorescent materials in Kupffer cells using scanning electron microscopy. Arch Histol Cytol 67(3):263–270
Kumar R, Nyk M, Ohulchanskyy TY, Flask CA, Prasad PN (2009) Combined optical and MR bioimaging using rare earth ion doped NaYF4 nanocrystals. Adv Funct Mater 19:853–859
Liv N, Zonnevylle AC, Narvaez AC, Effting APJ, Voorneveld PW, Lucas MS, Hardwick JC, Wepf RA, Kruit P, Hoogenboom JP (2013) Simultaneous correlative scanning electron and high-NA fluorescence microscopy. PLoS ONE 8(2), e55707
MacDonald L, Baldini G, Storrie B (2015) Does Super-resolution Fluorescence Microscopy obsolete previous microscopic approaches to protein colocalisation? Methods Mol Biol 1270:255–275
Mahfoud Z, Dijksman AT, Javaux C, Bassoul P, Baudrion A-L, Plain J, Dubertret B, Kociak M (2013) Cathodoluminescence in a scanning transmission electrom microscope: a nanometer-scale counterpart of photoluminescence for the study of II-VI quantum dots. J Phys Chem Lett 4:4090–4094
Morrison IEG, Dennison CL, Nishiyama H, Suga M, Sato C, Yarwood A, O’Toole PJ (2012) Atmospheric scanning electron microscope for correlative microscopy. Methods Cell Biol 111:307–324
Nishiyama H, Suga M, Ogura T, Maruyama Y, Koizumi M, Mio K, Kitamura S, Sato C (2010) Atmospheric scanning electron microscope observes cells and tissues in open medium through silicon nitride film. J Struct Biol 172:191–202
Nisman R, Dellaire G, Ren Y, Li R, Bazett-Jones DP (2004) Application of quantum dots as probes for correlative fluorescence, conventional and energy-filtered transmission electron microscopy. J Histochem Cytochem 52(1):13–18
Padman BS, Bach M, Ramm G (2014) An improved procedure for subcellular spatial alignment during live-cell CLEM. PLoS ONE 9(4), e95967
Patterson GH, Piston DW (2000) Photobleaching in two-photon excitation microscopy. Biophys J 78(4):2159–2162
Peddie CJ, Blight K, Melia C, Marrison J, Carzaniga R, Domart MC, O’Toole PJ, Larijani B, Collinson LM (2014) Correlative and integrated light and electron microscopy of in-resin GFP fluorescence, used to localise diacylglycerol in mammalian cells. Ultramicroscopy 143:3–14
Rapaport A, Milliez J, Bass M, Cassagno A, Jenssen H (2006) Review of the properties of Up-conversion phosphors for new emissive displays. J Disp Technol 2(1):68–78
Riwotzki K, Meyssamy H, Schnablegger H, Kornowski A, Haase M (2001) Liquid-phase synthesis of colloids and redispersible powders of strongly luminescing LaPO4:Ce, Tb nanocrystals. Angew Chem Int Ed 40:573–576
Salimian A, Fern GR, Withnall R, Silver J (2012) A novel method for the preparation of non-agglomerated nanometer sized particles of lanthanum phosphate phosphors utilising a high surface area support in the firing process. J Mater Chem 22:21529–21532
van Rijnsoever C, Oorschot V, Klumperman J (2009) A novel correlative light-electron microscopy (CLEM) method integrating live-cell imaging and immunolabelling of ultrathin cryosections. Nat Methods 5:973–980
Acknowledgments
This work was supported by BBSRC, EPSRC and MRC (awards BB/F004729/1 and MR/K01580X/1) with Cancer Research UK, the University of York, and JEOL UK. We thank Dr. Andrew Yarwood (JEOL UK) and Dr. David Stowe (Gatan UK) for advice and helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Robert Withnall was deceased.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Morrison, I.E.G., Samilian, A., Coppo, P. et al. Multicolour correlative imaging using phosphor probes. J Chem Biol 8, 169–177 (2015). https://doi.org/10.1007/s12154-015-0141-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12154-015-0141-5