
Abstract
Severe alcoholic hepatitis (SAH) is defined by modified Maddrey discriminant function ≥32 or Model for End-Stage Liver Disease (MELD) >21 and/or hepatic encephalopathy. It has a 3-month mortality rate ≥30–70 %. Patients with severe alcoholic hepatitis need combined, i.e., static (MELD score) and dynamic (Lille’s score), prognostication. Systemic inflammation and poor regeneration are hallmarks of SAH, rather than intrahepatic inflammation. SAH is characterized by dysregulated and uncontrolled systemic inflammatory response followed by weak compensatory antiinflammatory response that leads to increased susceptibility to infection and multiple organ failure. Massive necrosis of hepatocytes exceeds the proliferative capacity of hepatocytes. Liver progenitor cells proliferate to form narrow ductules which radiate out into the damaged liver parenchyma. Corticosteroids have been the standard-of-care therapy, albeit controversial. However, the recent Steroids or Pentoxifylline for Alcoholic Hepatitis (STOPAH) trial revealed that prednisolone was not associated with a significant reduction in 28-day mortality, with no improvement in outcomes at 90 days or 1 year. A paradigm shift from antiinflammatory therapy such as corticosteroids to liver regeneration treatment, e.g., granulocyte-colony stimulating factor, molecular targeted treatments, and fecal microbiota transplantation, for severe alcoholic hepatitis is taking place. Liver transplantation should be offered to select patients with severe alcoholic hepatitis who are nonresponsive to medical treatment.
Similar content being viewed by others
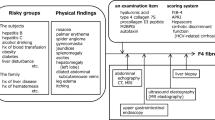

Introduction
Alcoholic liver disease (ALD) includes a broad spectrum of entities from simple steatosis, acute alcoholic hepatitis with or without underlying cirrhosis, to hepatocellular carcinoma as a complication of cirrhosis. Alcoholic hepatitis (AH) presents as mild or severe alcoholic hepatitis (SAH). The available therapeutic options for SAH are unsatisfactory. Appropriate diagnosis, prognostication, and better treatment options are essential to improve the survival rate.
Alcoholic hepatitis
Alcoholic hepatitis is defined as a syndrome of progressive inflammatory liver injury associated with decades of heavy alcohol use (chronic active alcohol abuse 60–80 g in males and 20–40 g in females) [1, 2]. The standard screening test for harmful alcohol use is the Alcohol Use Disorders Identification Test (AUDIT) [3]. The best biomarkers of alcohol use are γ-glutamyltransferase (GGT), carbohydrate-deficient transferrin (CDT), mitochondrial aspartate aminotransferase (AST), ethyl glucuronide (ETG), and phosphatidyl ethanol [4].
Clinical criteria for alcoholic hepatitis
The clinical criteria for alcoholic hepatitis are jaundice with duration <3 months, jaundice as first decompensating liver event, serum bilirubin >5 mg/dL; AST/alanine aminotransferase (ALT) >2:1, AST <500 IU/L, ALT <300 IU/L, and neutrophilic leukocytosis [1, 2]. In milder cases of alcoholic hepatitis, mild elevation of aspartate aminotransferase (AST) level may be the only diagnostic clue, without any symptoms [5].
Liver biopsy
Liver biopsy in AH is no longer routinely obtained, due to the high probability of accurate diagnosis of AH based on clinical assessment alone. Moreover, the facility for transjugular liver biopsy is absent in most clinics and community hospitals. However, liver biopsy should still be considered in select scenarios [4]:
-
Uncertainty about diagnosis of AH, due to incomplete or inconsistent history, or equivocal laboratory or imaging findings
-
Concern about coexisting liver injury (which may occur in 20 % of patients with ALD)
-
To assess the severity of AH before starting treatment, particularly corticosteroids
-
To assess for chronicity, or fibrosis, and thereby provide more accurate prognosis for the patient (although transient elastography may supplant biopsy for this indication in the future)
-
Enrollment in a clinical trial which requires liver biopsy to enhance patient homogenization
The approach for liver biopsy could be transjugular or percutaneous depending on clinician preference and resource availability. The transjugular route is generally recommended if the patient has ascites or significant coagulopathy, to reduce risk of complications (Class 1, Level C) [2].
The potential confounders of clinical SAH on liver biopsy (ASH) are age, obesity, nonalcoholic steatohepatitis, coexisting hepatitis C, iron overload, and primary biliary cirrhosis (due to presence of Mallory–Denk bodies) [6–8].
Noninvasive diagnostic modalities
Given the limitations and potential complications of liver biopsy, there is a need for alternative, noninvasive methods for diagnosis and assessment of disease severity of AH. Analysis of breath biomarkers, including volatile organic compounds and elemental gases, has been evaluated recently for diagnosis of AH [9]. Using receiver operating characteristic curve analysis, a model for diagnosis of AH was based on breath levels of trimethylamine and pentane (TAP). TAP scores of 36 or higher identified patients with AH (area under receiver operating characteristic curve 0.92) with 90 % sensitivity and 80 % specificity versus patients with acute decompensation or individuals without liver disease. Levels of exhaled trimethylamine moderately correlate with severity of AH. Larger studies are needed for validation.
Severe alcoholic hepatitis (SAH)
SAH is characterized clinically by rapid onset of jaundice and liver failure—coagulopathy, ascites, and hepatic encephalopathy. Its laboratory criteria are defined as modified Maddrey discriminant function (mDF) ≥32 or Model for End-Stage Liver Disease (MELD) ≥21 and/or hepatic encephalopathy [2]. SAH is associated with 1-month mortality of 30 % and 3-month mortality of ≥30–70 % [10, 11]. The prevalence of acute on chronic liver failure (ACLF) in patients with SAH varies from 65 to 95 % [12, 13].
Pathogenesis of AH (Fig. 1)
Animal model: The mouse model of chronic and binge ethanol feeding developed by Gao et al. [14] involves feeding mouse alcohol ad libitum for 10 days followed by acute bolus gavage.

Pathogenesis of alcoholic hepatitis. Lipopolysaccharide (LPS), toll-like receptor 4 (TLR4), reactive oxygen species (ROS), tumor necrosis factor α (TNF-α), tumor necrosis factor α receptor 1 (TNF-α R1), platelet-derived growth factor (PDGF), transforming growth factor β (TGF-β), nuclear factor-κB (NF-κB), extracellular signal regulated kinase (ERK), Toll/interleukin-1 receptor-domain containing adaptor inducing TGF-β (TRIF), monocyte chemotactic protein 1 (MCP-1), programmed cell death protein (PD1), T cell immunoglobulin (TCI), mucin protein 3 (TIM-3), damage-associated molecular patterns (DAMPs), interleukins 1, 6, 8, 10 (IL1, ILR1, IL6, IL8, IL10), cytochrome 2E1 (CYP2E1), 4-hydroxynonenol (4-HN), malondialdehyde (MDD), S-adenosylmethionine (SAM), fatty acid (FA), glutathione synthase (GSH), mitochondrial permeability transition pore (MTP)
Intestinal dysbiosis and changes in intestinal permeability: Small intestinal dysmotility and alteration in the bile pool result in excess Proteobacteria and reduced levels of Bacteroidaceae and Lactobacillus [15]. Ethanol intake disrupts intestinal tight junctions. Associated zinc deficiency impairs the intestinal barrier [16].
Acetaldehyde: The major pathway of alcohol metabolism is conversion to acetaldehyde by alcohol dehydrogenase. Acetaldehyde is metabolized by aldehyde dehydrogenase to acetic acid in mitochondria. Acetaldehyde is reactive and forms DNA and protein adducts [17]. These adducts act as neoantigens for the immune systems.
Microsomal ethanol-oxidizing system: This is the minor pathway for alcohol metabolism. The cytochrome CYP2E1 is induced in chronic alcoholism. It produces reactive oxygen species which cause lipid peroxidation, glutathione, and S-adenosylmethionine depletion.
Damage-associated molecular patterns (DAMPs): Hepatocytes damaged by DNA, protein, and lipid adducts release endogenous damage-associated molecular patterns. DAMPs result in proinflammatory cytokines, localization of immune cells to the site of injury, along with a collection of cytosolic protein complex machinery known as the “inflammasome.”
Immune system activation
Innate immune system: Hepatocyte-specific interferon regulatory factor 3 (IRF3) is essential for mitochondrial apoptosis pathway. Leaky small intestine with intestinal dysbiosis results in release of lipopolysaccharide (LPS)/endotoxin from Proteobacteria in the gut lumen, which enters the portal vein:
(a) LPS interacts with Toll-like receptor 4 (TLR 4) on Kupffer cells, which produces proinflammatory cytokines [tumor necrosis factor α (TNF-α), IL1, and IL17].
(b) Alcohol activates the complement pathway (C3, C4). Interaction of complement and Kupffer cells results in production of proinflammatory TNF-α and hepatoprotective (IL6) and cytoprotective (IL10) cytokines. TNF-α induces production of IL8 and CXCL1 by hepatocytes and hepatic stellate cells (HSCs). These produce chemokines for neutrophil recruitment.
Adaptive immunity: Lipid peroxidation products (malondialdehyde and 4-hydroxynonenol) serve as protein adducts—neoantigens which induce antibody formation. There is a resultant increase in T cell presence in the inflamed liver.
Immune paralysis: Programmed cell death protein (PD-1), T cell immunoglobulin, and mucin protein 3 (TIM-3) are inhibitory receptors on T-lymphocytes which are overexpressed in AH patients. This results in neutrophil phagocytic dysfunction, which causes severe bacterial infection and multiorgan failure [18].
Impaired regeneration in SAH: Usually, liver damage induces mature hepatocytes to proliferate and replace necrotic hepatocytes. In SAH, two-thirds of the parenchyma is involved with steatosis. There is marked centrilobular ballooning degeneration with clusters of neutrophils and periportal mononuclear infiltration [19]. This is due to decreased energy stores due to hypoxia and a shift in lipid metabolism, along with a shift in redox reactions caused by preferential oxidation of alcohol in zone 3 of the hepatic lobule by the microsomal ethanol-oxidizing system.
This exceeds the proliferative capacity of hepatocytes. The hepatic progenitor cells (HPCs) are thought to reside in the terminal bile ductules (canals of Hering). “Oval” cells are the descendants of the stem cells and are found in the portal and periportal regions in experimental animals within days of liver injury. These cells proliferate to form narrow ductules, which may stain positively for biliary cytokeratin CK19, and radiate out into the damaged parenchyma. There is also growing evidence that bone marrow stem cells may contribute to liver regeneration [20].
Assessment of severity and prognosis of alcoholic hepatitis
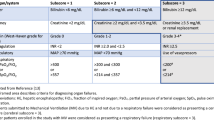
Assessment of disease severity and prognosis is critical for planning decisions regarding treatment of AH. Various scoring systems, such as the modified Maddrey’s discriminant function (mDF), Model for End-Stage Liver Disease (MELD), age, bilirubin, international normalized ratio (INR) , and creatinine (ABIC), and the Glasgow and Lille scoring systems are tested measures of disease severity (Table 1). The most recent scoring systems are as follows:
-
1.
Alcoholic hepatitis histologic score (AHHS)
This is a new scoring system. The AHHS was generated using the four histological features that independently predicted short-term survival, as detailed in Table 2 [21]. Presence of bridging fibrosis and cirrhosis is associated with poor outcome. This finding is not surprising given that extensive fibrosis leads to portal hypertension and favors related complications. Besides increasing intrahepatic resistance to blood flow, extensive fibrosis suggests more severe underlying liver disease and poor regenerative response in ACLF due to alcohol.
Presence of bilirubinostasis was associated with development of bacterial infection and sepsis. In fact, lipopolysaccharide (an important bacterial mediator that is markedly increased in AH) downregulates bile transporters in hepatocytes and causes cholestasis.
Neutrophilic infiltration and presence of megamitochondria identify patients with better outcomes.
-
2.
Combination of static and dynamic models
Louvet et al. [22] evaluated the prognostic value of combining static models for AH, such as mDF, MELD score, and ABIC score, with dynamic models, such as the Lille score. The MELD + Lille combination model predicted survival after 2 and 6 months significantly better than either the static or dynamic models alone and better than the mDF + Lille or ABIC + Lille models (p < 0.01) (Table 1).
-
3.
Biomarkers: Metabolomic profiling has identified elevated cytokines TNF-α, IL6, IL8, and IL15 in patients with SAH. Patients with serum IL6 levels >38.66 pg/mL had significantly decreased survival [23].
Treatment of alcoholic hepatitis
Treatment is based on disease severity and is response guided (Fig. 2).

Prognostication and response-guided therapy of alcoholic hepatitis. Corticosteroids (CS), alcoholic hepatitis (AH), liver transplant (LTX), granulocyte-colony stimulating factor (G-CSF), fecal microbiota transplantation (FMT)
Modalities of therapy
-
(a)
Abstinence
Abstinence from alcohol is the most important factor in predicting the outcome of acute episodes of AH as well as long-term survival. Incidence of recidivism after recovery from the first episode of AH varies from 10 to 70 %. Tools used to sustain abstinence include nonpharmacological and pharmacological methods:
-
1.
Nonpharmacological methods
These include outpatient motivational interviewing, cognitive behavioral therapy, and Alcoholics Anonymous (AA) attendance. In-patient therapy for alcoholism is required for patients who fail outpatient therapy, who have comorbid psychiatric disorders, and whose home situation is unstable.
-
2.
Drugs aiding abstinence
-
(a)
Naltrexone: Naltrexone exerts its principal pharmacological effects through blockade of the (μ) opioid receptor. Naltrexone also modifies the hypothalamic–pituitary–adrenal axis to suppress ethanol consumption. The usual dosage of naltrexone is 50 mg/day orally. Multiple meta-analyses of clinical trials for alcohol dependence found naltrexone to reduce alcohol consumption compared with placebo [24].
-
(b)
Acamprosate: This novel drug has structural similarities to the inhibitory neurotransmitter gamma-aminobutyric acid (GABA). Acamprosate has been shown to reduce withdrawal symptoms, including alcohol craving [25].
-
(c)
Baclofen: This GABA-β antagonist shows efficacy and safety in maintaining higher abstinence rates, longer duration of abstinence, and improved liver function tests in patients with alcoholic liver disease in randomized controlled trials (RCTs) [26]. The dosage is 5–10 mg orally three times per day .
-
(a)
Newer tests used to monitor abstinence
-
(a)
Breath test using a portable analyzer to detect breath alcohol content [27],
-
(b)
Ethyl glucuronide in urine [28] detects alcohol intake within the last 3 days,
-
(c)
Ethyl glucuronide in hair [29] detects intake of alcohol within the last few months with sensitivity and specificity of 92 and 91 %, respectively.
(b) Nutrition
Alcoholics commonly have significant protein–calorie malnutrition, along with deficiencies in a number of vitamins and trace minerals [30]. Severity of malnutrition has been shown to correlate with disease severity and survival [31]. In a recent study by Moreno et al. [32], a greater proportion of patients with daily calorie intake less than 21.5 kcal/kg/day died [65.8 %; 95 % confidence interval (CI) 48.8–78.4 % ] than patients with higher calorie intake (33.1 %; 95 % CI 23.1–43.4 %) (p < 0.001). The conclusion drawn from this negative study are:
-
First, each patient should be assessed for nutritional status on admission. The optimal nutritional evaluation is done by measuring sarcopenia [33], generally defined as a reduction in muscle mass two standard deviations below the healthy young adult mean. New sarcopenia cutoff values for patients with cirrhosis have been reported recently (L3 vertebra skeletal muscle index ≤42 cm2/m2 for women and ≤50 cm2/m2 for men) [34]. Assessment of sarcopenia by computed tomography (CT) or magnetic resonance imaging (MRI) is currently the gold standard for evaluating sarcopenia. Second, oral nutrition should be strongly encouraged to achieve a minimum of 21.5 kcal/kg per day, in line with advice provided in international guidelines.
-
However, in patients who fail to achieve this level of oral intake, careful consideration should be given to the benefit–risk ratio before instituting nasogastric or parenteral nutrition.
-
Randomised studies specifically evaluating (a) the role of supplemental enteral versus parenteral nutrition and (b) macronutrient composition with special attention to protein and lipid contents are required.
-
Parenteral feeds, which included amino acid supplementation through peripheral intravenous lines, showed promise in the 1980s but may now need to be evaluated in rigorously designed studies [35].
Enteral nutrition
-
Components: protein 1.5 g/kg/day, energy 40 kcal/kg/day, B-complex vitamins daily
-
Frequency: six meals a day including a nighttime snack
-
Oral branched-chain amino acids [35]
Treatment of SAH
Standard pharmacotherapy
-
1.
Corticosteroids (CS): This has been the most extensively studied intervention in patients with SAH. The rationale for use of CS is to reduce the immune and proinflammatory cytokine response, which is greatly increased in SAH and is responsible for liver injury [1, 36, 37]. Oral prednisolone 40 mg daily or intravenous methylprednisolone 32 mg daily, for 4 weeks, is the standard therapy. The recent STOPAH trial, conducted in 50 centers in the UK [38], did not find a significant benefit of CS in SAH. A limitation of this trial is the lack of histopathological confirmation and low mortality (19 %) in the placebo arm compared with 36 % in previous trials [1].
In another very recent study, data were taken from 1974 patients originating from nine RCTs, then three different meta-analyses were performed: CS versus placebo (n = 6 trials), CS versus pentoxifylline (PTX) (n = 2), and CS + PTX versus CS + placebo (n = 3). The study concluded that CS improved 28-day survival in patients with SAH with higher response rate compared with PTX and placebo. This treatment benefit was sustained until the end of therapeutic period. The combination of CS and PTX did not add any additional effect [39].
-
2.
Pentoxifylline (PTX)
Pentoxifylline is an oral phosphodiesterase inhibitor that also inhibits production of TNF-α, among other proinflammatory cytokines. It is given at dosage of 400 mg thrice a day for 28 days. In a pivotal study by Akriviadis on 101 patients with SAH [40], it was associated with survival benefit of 50 % and decrease in hepatorenal syndrome (HRS) in the treatment group. In a study including 50 patients at our center [41], we found reduction in short-term mortality in the pentoxifylline group compared with controls (20 versus 40 %). A meta-analysis of five RCTs failed to show any survival benefit with PTX in patients with SAH [42]. The STOPAH study concluded that PTX is no better than placebo in SAH [38].
-
3.
Sequential therapy (CS followed by PTX)
In the study by Louvet et al. [43], 29 steroid nonresponders were switched to PTX for 28 days and compared with 58 nonresponders treated with CS for 28 days. No improvement in 2-month survival was noted in the sequential therapy group compared with the CS group (35.5 versus 31 %).
-
4.
Combination therapy (PTX in combination with CS)
Combination of CS and PTX appears to be an attractive strategy on the basis of their potential synergistic action [44]. To study this, we conducted a randomized placebo controlled trial [10] comparing combination of CS plus PTX versus CS alone for 28 days. On intention-to-treat analysis, the 6-month survival and incidence of HRS were similar in the two arms. A similar trial from Europe reported concordant results [44].
Other therapies
-
1.
Antioxidants
-
(a)
N-Acetylcysteine (NAC): In a recent randomized trial on 174 patients with SAH, use of NAC + CS versus CS alone improved patient survival at 1 month with lower incidence of sepsis. However, there was no survival advantage at 6 months [45]. An ongoing study combines CS with NAC to augment CS function [46].
-
(a)
-
(b)
Metadoxine: Addition of metadoxine to glucocorticoid treatment improved short-term survival of patients with SAH and diminished development or progression of encephalopathy and hepatorenal syndrome [47]. Moreover, metadoxine improved the 3- and 6-month survival rates in patients with SAH [48].
-
2.
Tumor necrosis factor α (TNF-α) molecular inhibitors
TNF-α activates inflammatory pathways besides stimulating genes for hepatocyte growth factor production and regeneration. Parenteral TNF-α inhibitors have been tried in treatment of SAH. However, due to lack of efficacy and significant increase in infections, use of agents such as infliximab and etanercept should be confined to clinical trials only [49].
-
3.
Miscellaneous
Cochrane reviews showed no benefit of colchicine [50], anabolic steroids [51], S-adenosylmethionine (SAMe) [52], and prophylthiouracil [53].
-
4.
Granulocytapheresis was useful in six patients with SAH (five were CS nonresponders) [54].
-
5.
Molecular adsorbent recirculating system (MARS) did not improve survival [55].
Newer therapies
Just as there is a threshold for initiating steroids (mDF score >32 or MELD >21), there may also be a ceiling beyond which medical therapies aimed at decreasing the inflammatory cascade may cause more harm than benefit. One study suggested that patients with mDF >54 were at higher mortality risk from use of CS than from not being treated. Results of the COrticosteroids plus PEntoxifylline in severe alcoholic hepatitis (COPE) study [10] supported this fact, as the 6-month survival in the CS treatment group was just 23.5 % compared with 65 % reported by Louvet et al. [56] (baseline mDF-85 versus 57). Therefore, there is a pressing need for newer therapies to treat these extremely sick SAH patients.
-
(a)
Liver regeneration treatment:
Granulocyte-colony stimulating factor (G-CSF)
Dubuquoy et al. [57] recorded absence of liver regeneration cytokines TNF-α and IL6 in explant livers of 16 patients nonresponsive to CS. G-CSF promotes mobilization of bone marrow stem cells which populate liver and differentiate into hepatic cells. They improve neutrophil dysfunction and overcome immune paralysis in SAH [58–60]. The neutrophils also secrete cytokines that stimulate liver regeneration (Fig. 3). Singh et al. [11] used G-CSF at dosage of 5 mcg/kg B.I.D. for 5 days, comparing it with PTX. G-CSF resulted in mobilization of CD34+ cells, decreased infections, and significantly improved mDF and survival at 3 months in SAH patients, and is safe [60]. There are two ongoing studies on the efficacy and safety of G-CSF in patients with severe alcoholic hepatitis with null or partial response to CS [61] and in CS refractory patients [62]. G-CSF is given at 5 µg/kg daily for 5 days followed by once every 3 days for a total of 12 doses.

Mechanism of action of granulocyte-colony stimulating factor
-
(b)
Molecular targeted therapies
These are based on the pathogenesis of AH (Fig. 4):
Fig. 4 
Molecular targeted therapies; abbreviations: Toll-like receptor 4 (TLR4) receptors, interleukin 2 receptor (IL2R) antagonist (anakinra), Kupffer cells (KCs), macrophages (MAC), neutrophils (NEUT), antibody (Ab), interleukin (IL)17, interleukin (IL)22, HSC hepatic stellate cell, MCP monocyte chemotactic protein, hepatocytes (HEP), tumor necrosis factor (TNF) receptor 1, farnesoid X receptor (FXR)

-
I.
Strengthening the leaky gut barrier, reversal of gut dysbiosis, and decreasing luminal and portosystemic endotoxemia (Table 3).
(a) Bovine colostrum: Bovine colostrum is rich in proteins, immunoglobulins (20–30 % IgG, an anti-endotoxin antibody), lactoferrin, and growth factors. Lactoferrin is converted to lactoferricin B, which kills gut Gram-negative bacilli. IgG interacts with mucosa-associated lymphoid tissue of the leaky gut and normalizes leaky gut permeability, decreasing entry of lipopolysaccharide/endotoxin into the portal circulation [63]. Lactoferrin binds to lipid-A part of the lipopolysaccharide. Lactoferrin and IgG act synergistically in neutralizing luminal and portal venous endotoxemia. The subsequent cascade of proinflammatory cytokines including TNF-α and interleukins 6 and 8 is decreased [64]. A study on 25 SAH patients given corticosteroids and bovine colostrum at our center showed very encouraging results [65].
(b) Hyperimmune bovine colostrum: The hypothesis is that oral administration of hyperimmune bovine colostrum (Imm124-E) enriched with anti-LPS antibodies will reduce endotoxemia and improve SAH pathophysiological and clinical parameters. This is under study in the Translational Research and Evolving Alcoholic Hepatitis Treatment (TREAT) consortium study [66].
(c) Fecal microbiota transplantation (FMT): Recently, FMT has been successfully used in treatment of life-threatening infections with Clostridium difficile. Gut bacteria dysbiosis in the small intestine, such as increased Proteobacteria and decreased Bacteroidetes, are actively involved in the pathogenesis of alcoholic hepatitis. FMT through nasogastric tube might have a potential role in management of SAH [67]. In a more recent pilot study, 1 week of FMT was effective and safe in SAH patients and improved indices of liver disease severity and survival at 1 year. Improvement in liver function and survival could have been due to improvement in sepsis, nutritional rehabilitation, and abstinence. New species from the donor, which are less pathogenic and beneficial, coexist with preexisting bacterial communities of the recipient. It is likely that the latter are substantially modified by the donor species [68].
-
II.
Deactivation of liver innate immunity
Kupffer cells: Endotoxin activates Kupffer cells (KCs), which produce proinflammatory cytokine interleukin (IL)1β. IL1β further recruits thymocyte (Th)17 cells, which activate neutrophils, hepatic stellate cells, and necrosis of hepatocytes. A study is evaluating a combination of anakinra, an IL1β receptor antagonist, given by 100-mg subcutaneous injection daily for 14 days, pentoxifylline 400 mg orally three times daily for 28 days, and zinc sulfate 220 mg orally for 180 days versus methylprednisolone 32 mg IV for 28 days [69].
-
III.
Attenuation of hepatocellular necrosis, apoptosis, and fibrosis
(a) Tumor necrosis factor alpha (TNF-α): This proinflammatory cytokine is released by endotoxin-stimulated Kupffer cells and macrophages in liver, which also stimulates hepatocyte regeneration. Total inhibition of TNF-α by infliximab is harmful, preventing hepatocellular regeneration and causing septicemia and death [49].
(b) Emricasan: Caspases are death-induction molecules situated downstream from TNF-α in the hepatocyte injury signaling cascade. Emricasan, a pancaspase inhibitory compound, has been used in treatment for SAH at dosage of 25 mg BID for 28 days [74]. However, the trial was terminated due to high systemic blood levels exceeding levels in toxicology studies.
(c) Human interleukin 22 (IL22): This cytokine is hepatoprotective. F-652, a recombinant fusion protein containing IL22 and human immunoglobulin G2 (IgG2)-Fc produced in Chinese hamster ovary (CHO) cells in serum-free culture, is being used in a safety/efficacy study to treat patients with alcoholic hepatitis [75].
(d) Obeticholic acid (OCA): OCA has been found to be effective in patients with moderately severe alcoholic hepatitis. OCA is a bile acid analog and a potent first-in-class agonist of farnesoid X receptor (FXR). The major function of FXR is to suppress bile acid biosynthesis from cholesterol and regulate hepatic triglyceride levels. The dosage of OCA is 10 mg once a day orally for 6 weeks [76].
(e) GS-4997 (simtuzumab): This monoclonal antibody is selective for lysyl oxidase-like-2, an extracellular matrix enzyme that promotes fibrosis via cross-linkage of collagen fibers. Gilead is evaluating simtuzumab for treatment of fibrosis in patients with SAH [77].
Liver transplantation
SAH patients listed for liver transplant (LT) have to undergo exhaustive psychosocial evaluation. The Lille group study recorded that the 6-month survival of transplanted patients was higher than for those not transplanted [78]. In a study from the USA, 9 (9.6 %) of 94 refractory SAH patients underwent early LT, accounting for 3 % of all adult LT during the study period. The 6-month survival rate was significantly higher among those receiving early LT compared with matched controls. Eight recipients were alive at a median of 735 days, with one alcohol relapse. Thus, early LT for severe AH can achieve excellent clinical outcomes with low impact on the donor pool and low rates of alcohol relapse in highly selected patients [79].
Conclusions
-
(A)
Prognostication
The combination of MELD + Lille is more accurate than either score alone.
-
(B)
Therapy
-
1.
Corticosteroids are the gold standard of therapy. However, they are discouraged at mDF >54.
-
2.
Enteral nutrition. Nutritional assessment at onset is essential. Newer trials assessing intravenous amino acids in nutritional supplementation of SAH are needed.
(C) SAH patients where CS are refractory or contraindicated
-
1.
G-CSF
-
2.
FMT or option for inclusion in new trials in addition to G-CSF
-
3.
CS + IV NAC for 2–4 weeks
-
4.
Early liver transplantation in abstinence-motivated patients with strong psychosocial support
Future perspectives for research in management of SAH
-
(a)
Paradigm shift in therapy of SAH: Anti-inflammatory therapies, e.g., CS, will probably be replaced by therapies promoting liver regeneration such as G-CSF, molecular targeted therapies, and reversal of gut dysbiosis by fecal microbiota transplantation.
-
(b)
Molecular drivers of fibrosis: In SAH with cirrhosis, molecular drivers of fibrosis need to be identified. Newer antifibrogenic agents need to be studied.
References
Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med 2009;2009(360):2758–2769
O’Shea RS, Dasarathy S, McCullough AJ. The Practice Guideline Committee of the American Association for the Study of Liver Diseases and the Practice Parameters Committee of the American College of Gastroenterology. Alcoholic liver disease. Hepatology 2010;2010:307–328
Saunders JB, Aasland OG, Babor TF, de la Fuente JR, Grant M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO collaborative project on early detection of person with harmful alcohol consumption-II. Addiction 1993;1993(88):791–804
Childer RE, Joseph AH. Diagnosis of alcoholic hepatitis liver disease. Clin Liver Dis 2016;2016(20):457–471
Heuman DM, Hung PD. Alcoholic hepatitis. Drug and disease. Gastroenterology. http://emedicine.medscape.com/article/170539-overview
Crabb DW, Bataller R, Chalasani NP, Kamath PS, Lucey M, Mathurin P, et al. Standard definitions and common data elements for clinical trials in patients with alcoholic hepatitis: recommendation from the NIAAA alcoholic hepatitis consortia. Gastroenterology 2016;150(4):785–790
Kato M, Kato S, Horiuchi S, Nagai K, Horie Y, Hayashi K. Mallory bodies in hepatocytes of alcoholic liver disease and primary biliary cirrhosis contain-(carboxymethyl) lysine-modified cytokeratin, but not those in hepatic carcinoma cells. Yonago Acta Med 2006;49:83–92
EASL Clinical Practical Guidelines. Management of alcoholic liver disease. J Hepatol 2012;57:399–420
Hanouneh IA, Zein NN, Cikach F, Dababneh L, Grove D, Alkhouri N, et al. The breath prints in patients with liver disease identify novel breath biomarkers in alcoholic hepatitis. Clin Gastroenterol Hepatol 2014; 12(3): 516–523
Sidhu SS, Goyal O, Singla P, Sood A, Chhina RS, Soni RK. Corticosteroid plus pentoxifylline is not better than corticosteroid alone for improving survival in severe alcoholic hepatitis (COPE Trial). Dig Dis Sci 2012;57(6):1664–1671
Singh V, Sharma KA, Narasimhan RL, Bhalla A, Sharma N, Sharma R. Granulocyte colony-stimulating factor in severe alcoholic hepatitis: a randomized pilot study. Am J Gastroenterol 2014;2014(109):1417–1423
Dominguez M, Rincon D, Abraldes JG, Miquel R, Colmenero J, Bellot P, et al. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol 2008;2008(103):2747–2756
O’Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Am J Gastroenterol 2010;2010(105):14–32
Arteel GE. Build a better mouse model, and the world will beat a path to your door. Hepatology 2013;58(5):1526–1528
Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013;58(5):949–955
Zhong W, McClain CJ, Cave M, Kang YJ, Zhou Z. The role of zinc deficiency in alcohol-induced intestinal barrier dysfunction. Am J Physiol Gastrointest Liver Physiol 2010;298(5):G625–G633
Setshedi M, Wands JR, Monte SM. Acetaldehyde adducts in alcoholic liver disease. Oxid Med Cell Longev 2010; 3(3): 178–185
Dunn W, Shah VH. Pathogenesis of alcoholic liver disease. Clin Liver Dis 2016;2016(20):445–456
Alpert L, Hart J. The pathology of alcoholic liver disease. Clin Liver Dis 2016;2016(20):473–489
Vessey Carina J, Pauline M, De La Hall M. Hepatic stem cells: a review. Pathology 2001;33:130–141
Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Rojo AD, Louvet A, et al. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology 2014; 146(5):1231–1239
Louvet A, Labreuche J, Artru F, Boursier J, Kim DJ, O’Grady J, et al. Combining data from liver disease scoring systems better predicts outcomes of patients with alcoholic hepatitis. Gastroenterology 2015;149(2): 398–406
Rachakonda V, Gabbert C, Raina A, Li H, Malik S, DeLany JP, et al. Stratification of risk of death in severe acute alcoholic hepatitis using a panel of adipokines and cytokines. Alcohol Clin Exp Res 2014;2014(38):2712–2721
Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, et al. Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta-analysis. JAMA 2014; 311:1889
Mason BJ. Acamprosate in the treatment of alcohol dependence. Expert Opin Pharmacother 2015;2005(6):2103–2115
Frazier TH, Stocker AM, Kershner NA, Marsano LS, McClain CJ. Treatment of alcoholic liver disease. Therap Adv Gastroenterol 2011; 4(1): 63–81
Kaisdotter A, Kron J, Castren M, Athlin AM, Hok B, Wiklund L. Assessment of the breath alcohol concentration in emergency care patients with different level of consciousness. Scand J Trauma Resusc Emerg Med 2015; 23:11
Piano SM, Rosi S, Cavallin M, Romano A, Gola E, Marchioro L, et al. Urinary ethyl glucuronide improves the detection of alcohol consumption in liver transplantation candidates and recipients. Hepatology 2012;56:977A
Sterneck MA, Staufer K, Schulz KH, Nashan B. Ethyl glucuronide determination in hair for evaluation of long-term alcohol abstention in liver transplant candidates. Hepatology 2012;56:493A
Mezey E. Interaction between alcohol and nutrition in the pathogenesis of alcoholic liver disease. Semin Liver Dis 1991;1991(11):340–348
Mendenhall CL, Anderson S, Weesner RE, Goldberg SJ, Crolic KA. Protein-calorie malnutrition associated with alcoholic hepatitis. Veterans Administration Cooperative Study Group on Alcoholic Hepatitis. Am J Med 1984;1984(76):211–222
Moreno C, Deltenre P, Senterre C, Louvet A, Gustot T, Bastens B, et al. Intensive enteral nutrition is ineffective for patients with severe alcoholic hepatitis treated with corticosteroids. Gastroenterology 2016;2016(150):903–910
Sinclair M, Gow PJ, Grossmann M, Angus PW. Review article: sarcopenia in cirrhosis-aetiology, implications and potential therapeutic interventions. Aliment Pharmacol Ther 2016;43(7):765–777
Montano-Loza AJ, Meza-Junco J, Prado CMM, Tandon P, Bain VG, Ma M, et al. New cutoff values for sarcopenia for predicting 6-month mortality in cirrhotic patients. J Hepatol 2013;58(Suppl):S95
Puri P, Thurz M. Intensive enteral nutrition in alcoholic hepatitis: more food for thought. Gastroenterology 2016; 150(4): 803–805
Taieb J, Mathurin P, Elbim C, Cluzel P, Arce-Vicioso M, Bernard B, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol 2000;2000(32):579–586
Han J, Thompson P, Beutler B. Dexamethasone and pentoxifylline inhibit endotoxin-induced cachectin/tumor necrosis factor synthesis at separate points in the signaling pathway. J Exp Med 1990;1990(172):391–394
Thursz M, Richardson P, Allison M, Austin A, Bowers M, Christopher P, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015;2015(372):1619–1628
Thursz MR, Louvet A, Kim DJ, Labreuche J, Atkinson SR, Sidhu S, et al. Corticosteroids are the only remaining pharmacological option for severe alcoholic hepatitis: a meta-analysis of individual data on 1974 patients. In Paper presented at AASLD
Akriviadis E, Botla R, Briggs W, Han S, Reynolds T, Shakil O. Pentoxifylline improves short-term survival in severe acute alcoholic hepatitis: a double-blind, placebo-controlled trial. Gastroenterology 2015;2000(119):1637–1648
Sidhu SS, Goyal O, Singla M, Bhatia KL, Chhina RS, Sood A. Pentoxiphylline in severe alcoholic hepatitis: a prospective, randomized trial. J Assoc Phys India 2012;2011(69):20–22
Whitfield K, Rambaldi A, Wetterslev J, Gluud C. Pentoxifylline for alcoholic hepatitis. Cochrane Database Syst Rev 2009; CD007339
Louvet A, Diaz E, Dharancy S. Early switch to pentoxifylline in patients with severe alcoholic hepatitis is inefficient in non-responders to corticosteroids. J Hepatol 2008;2008(48):465–470
Mathurin P, Louvet A, Duhamel A, Nahon P, Carbonell N, Boursier J, et al. Prednisolone with vs without pentoxifylline and survival of patients with severe alcoholic hepatitis. A randomized clinical trial. JAMA 2013;310(10):1033–1041
Nguyen-khac E, Theevenot T, Benferhat S, Goria O, Chatelain D, Tramier B, et al. Glucocorticoids plus N-acetyl cysteine in severe alcoholic hepatitis. N Engl J Med 2011;365:1781–1789
Centre Hospitalier Universitaire, Amiens. Treatment of Severe Alcoholic Hepatitis with Corticoids Plus N Acetyl Cysteine Versus Corticoids Alone (HAA-NAC). https://clinicaltrials.gov/ct2/show/NCT00863785
Higuera-de la Tijera F, Servin-Caamano AI, Cruz-Herrera J, Serralde-Zuniga AE, Abdo-Francis JM, Gutierrez-Reyes G, et al. Treatment with metadoxine and its impact on early mortality in patients with severe alcoholic hepatitis. Ann Hepatol 2014;13(3):343–352
Higuera-de la Tijera F, Servin-Caamano AL, Serralde-Zuniga AE, Herrera JC, Torres EP, Abdo-Francis AM, et al. Metadoxine improves the three- and six-month survival rates in patients with severe alcoholic hepatitis. World J Gastroenterol 2015; 21(16): 4975–4985
Naveau S, Chollet-Martin S, Dharancy S, Jouet P, Piquet MA, Davion T, et al. A double-blind randomized controlled trial of infliximab associated with prednisolone in acute alcoholic hepatitis. Hepatology 2004;2004(39):1390–1397
Rambaldi A, Gluud C. Colchicine for alcoholic and non-alcoholic liver fibrosis and cirrhosis (review). Cochrane Database Syst Rev 2001;(3):CD002148
RambaldiAL, Iaquinto G, Gluud C. Anabolic-androgenic steroids for alcoholic liver disease. Cochrane Database Syst Rev 2003;(1):CD003045
Mato JM, Camara J, Fernandez J, Caballeria L, Coll S, Caballero A, et al. S-Adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo-controlled, double-blind, multicenter clinical trial. J Hepatol 1999;1999(30):1081–1089
Orrego H, Blake JE, Blendis LM, Compton KV, Israel Y. Long-term treatment of alcoholic liver disease with propylthiouracil. N Engl J Med 1987;1987(317):1421–1427
Morris JML, Dickson S, Neilson M, Hodgins P, Forrest EH. Granulocytapheresis in the treatment of severe alcoholic hepatitis: a case series. Eur J Gastroenterol Hepatol 2010;22(4):457–460
Jalan R, Sen S, Steiner C, Kapoor D, Alisa A, Williams R. Extracorporeal liver support with molecular adsorbents recirculating system in patients with severe acute alcoholic hepatitis. J Hepatol 2003(54):24–31
Louvet A, Naveau S, Abdelnour M, Ramond MJ, Diaz E, Fartoux L, et al. The Lille model: a new tool for therapeutic strategy in patients with severe alcoholic hepatitis treated with steroids. Hepatology 2007;2007(45):1348–1354
Dubuquoy L, Louvet A, Lassailly G, Truant S, Boleslawski E, Artru F, et al. Progenitor cell expansion and impaired hepatocyte regeneration in explanted livers from alcoholic hepatitis. Gut 2015;2015(64):1949–1960
Tato CM, Cua DJ. SnapShot: cytokines IV. Cell 2008; 132:1062
Rolas L, Makhezer N, Hadjoudj S, El-Benna J, Djerdjouri B, Elkrief L, et al. Inhibition of mammalian target of rapamycin aggravates the respiratory burst defect of neutrophils from decompensated patients with cirrhosis. Hepatology 2013;2013(57):1163–1671
Moreau R, Rautou PE. G-CSF therapy for severe alcoholic hepatitis: targeting liver regeneration or neutrophil function? Am J Gastroenterol 2014;2014(109):1424–1426
Kim DJ. Efficacy and Safety of Granulocyte-Colony Stimulating Factor in Patients with Severe Alcoholic Hepatitis with Null or Partial Response to Steroid: A Randomized, Double-Blind, Placebo-Controlled, Nationwide Multi-Center Study. https://clinicaltrials.gov/ct2/show/NCT02442180
Shasthry SM. Efficacy of G-CSF in the Management of Steroid Non-Responsive Severe Alcoholic Hepatitis. A Double Blind Randomized Control Trial. https://clinicaltrials.gov/ct2/show/NCT01820208
Struff WG, Sprotte G. Bovine colostrum as a biologic in clinical medicine: a review—Part II. Int J Clin Pharmacol Ther 2008;46:211–225
Kelly GS. Bovine colostrums: a review of clinical uses. Altern Med Rev 2003;8:378–394
Sidhu S, Goyal O, Kishore H. Corticosteroids and Bovine Colostrum in Treatment of Alcoholic Hepatitis ‘In Extremis’: A Pilot Study. In Paper Presented at Emerging Trends Conference in Alcohol Hepatitis. JW Marriott Miami, 22–23 January 2016
Virginia Commonwealth University. National Institute on Alcohol Abuse and Alcoholism (NIAAA). Safety and Efficacy of IMM 124-E for Patients with Severe Alcoholic Hepatitis (TREAT). https://clinicaltrials.gov/ct2/show/NCT01968382
Institute of Liver and Biliary Sciences, India. Pentoxyphilline Versus Fecal Microbiota Therapy in Severe Alcoholic Hepatitis. https://clinicaltrials.gov/ct2/show/NCT02458079
Philips CA, Pande A, Shasthry SM, Jamwal KD, Khillan V, Chandel SS, et al. Healthy donor fecal microbiota transplantation in steroid ineligible severe alcoholic hepatitis—a pilot study. Clin Gastroenterol Hepatol 2016;. doi:10.1016/j.cgh.2016.10.029
Efficacy Study of Anakinra, Pentoxifylline, and Zinc Compared to Methylprednisolone in Severe Acute Alcoholic Hepatitis. https://clinicaltrials.gov/ct2/show/NCT01809132
University Hospital, Lille. Efficacy of Antibiotic Therapy in Severe Alcoholic Hepatitis Treated with Prednisolone (AntibioCor). https://clinicaltrials.gov/ct2/show/NCT02281929
University of Texas South Western Medical Centre. Novel Therapies in Moderately Severe Acute Alcoholic Hepatitis (NTAH-Mod).https://clinicaltrials.gov/ct2/show/NCT01922895
Hospital Universitari Vall d’Hebron Research Institute. Effects of Rifaximin in Patients with Acute Alcoholic Hepatitis (RIFA-AAH). https://clinicaltrials.gov/ct2/show/NCT02116556
Helsinki University. Randomised Open-Label Multicentre Study Evaluating Ciprofloxacin in Severe Alcoholic Hepatitis. https://clinicaltrials.gov/ct2/show/NCT02326103
Conatus Pharmaceuticals Inc. Study of IDN-6556 in Patients with Severe Alcoholic Hepatitis and Contraindications to Steroid Therapy (AH). https://clinicaltrials.gov/ct2/show/NCT01912404.
Mayo Clinic. Use of F-652 in Patients with Alcoholic Hepatitis (TREAT 008). https://clinicaltrials.gov/ct2/show/NCT02655510.
Indiana University. Trial of Obeticholic Acid in Patients with Moderately Severe Alcoholic Hepatitis (TREAT 002). https://clinicaltrials.gov/ct2/show/NCT02039219.
Gilead Sciences. GS-4997 in Combination with Prednisolone Versus Prednisolone Alone in Participants with Severe Alcoholic Hepatitis. https://clinicaltrials.gov/ct2/show/NCT02854631.
Brown RS Jr. Transplantation for alcoholic hepatitis time to rethink the 6-month rule. N Engl J Med 2011;2011(365):1836–1838
Mathurin P, Moreno C, Samuel D, Dumortier J, Salleron J, Durand F, et al. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med 2011;2011(365):1790–1800
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Sandeep Singh Sidhu, Omesh Goyal, Harsh Kishore, and Simran Sidhu declare that they have no conflicts of interest.
Informed consent
None.
Financial support
None.
Additional information
The work has been done in Dayanand Medical College and Hospital.
Rights and permissions
About this article

Cite this article
Sidhu, S.S., Goyal, O., Kishore, H. et al. New paradigms in management of alcoholic hepatitis: a review. Hepatol Int 11, 255–267 (2017). https://doi.org/10.1007/s12072-017-9790-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12072-017-9790-5