
Abstract
Background
The hypoxia-inducible factor-1α (HIF-1α)/vascular endothelial growth factor (VEGF)/VEGF receptor subtype 2 (VEGFR-2) pathway has been implicated in ischemia/reperfusion injury. The aim of this study was to clarify whether whole-body hypothermic targeted temperature management (HTTM) inhibits the HIF-1α/VEGF/VEGFR-2 pathway in a swine model of cardiac arrest (CA) and cardiopulmonary resuscitation (CPR).
Methods
Twenty-four domestic male Beijing Landrace pigs were used in this study. CA was electrically induced with ventricular fibrillation and left untreated for 8 min. Return of spontaneous circulation (ROSC) was achieved in 16 pigs, which were randomly assigned either to normothermia at 38 °C or to HTTM at 33 °C (each group: n = 8). HTTM was intravascularly induced immediately after ROSC. The core temperature was reduced to 33 °C and maintained for 12 h after ROSC. The serum levels of HIF-1α, VEGF, VEGFR-2, and neuron-specific enolase (NSE) were measured with enzyme immunoassay kits 0.5, 6, 12, and 24 h after ROSC. The expression of HIF-1α, VEGF, and VEGFR-2 in cerebral cortical tissue was measured by RT-PCR and Western blot analysis 24 h after ROSC. Neurological deficit scores and brain cortical tissue water content were evaluated 24 h after ROSC.
Results
The serum levels of HIF-1α, VEGF, and VEGFR-2 were significantly increased under normothermia within 24 h after ROSC. However, these increases were significantly reduced by HTTM. HTTM also decreased cerebral cortical HIF-1α, VEGF, and VEGFR-2 mRNA and protein expression 24 h after ROSC (all p < 0.05). HTTM pigs had better neurological outcomes and less brain edema than normothermic pigs.
Conclusion
The HIF-1α/VEGF/VEGFR-2 system is activated following CA and CPR. HTTM protects against cerebral injury after ROSC, which may be part of the mechanism by which it inhibits the expression of components of the HIF-1α/VEGF/VEGFR-2 signaling pathway.
Similar content being viewed by others

Introduction
Brain injury is the leading cause of mortality and morbidity among patients resuscitated from cardiac arrest (CA) [1, 2]. The derived neurological deficits result from whole-body ischemia/reperfusion damage after CA and cardiopulmonary resuscitation (CPR) [3]. Therapeutic hypothermia or hypothermic targeted temperature management (HTTM, 32–36 °C) has emerged as a proven strategy to minimize secondary brain damage in survivors of CA and has become the recommended treatment by the American Heart Association [4]. Hypothermia protects against CA- and resuscitation-induced brain injury and improves neurological outcome in CA patients and animals [5, 6]; these effects are multifactorial and may involve in the inhibition of inflammatory reactions, decrease in oxidative stress, attenuation of blood–brain barrier disruption, and inhibition of early brain edema [7,8,9]. However, the precise mechanism of this protection is largely unknown.
Hypoxia-inducible factor (HIF)-1, an essential regulator of oxygen homeostasis, is upregulated in response to hypoxia–ischemia. HIF-1 activity depends on the availability and activity of the subunit HIF-1 alpha (HIF-1α) [10]. HIF-1α is an important transcription factor implicated in ischemia–reperfusion brain injury [11, 12]. Vascular endothelial growth factor (VEGF), a well-characterized target gene of HIF-1α, can promote vascular permeability, disrupt the blood–brain barrier (BBB), and induce cerebral edema [13,14,15,16,17,18]. In addition, VEGF bound to VEGF receptor subtype 2 (VEGFR-2) and induced BBB disruption [16, 19,20,21]. Therefore, the HIF-1α/VEGF/VEGFR-2 pathway seems to be closely associated with ischemia-induced BBB breakdown and cerebral edema. Previous studies have shown that early inhibition of HIF-1α and VEGF expression can attenuate neuronal deficits and cerebral edema caused by ischemia/reperfusion injury [12, 22]. We speculated that HTTM attenuates neurological dysfunction and that cerebral edema may inhibit the expression of components of the HIF-1α/VEGF/VEGFR-2 axis in a pig model of global cerebral ischemia following CA and CPR.
Based on this background, our study aimed to test the hypothesis that HTTM attenuates postresuscitation brain injury and reduces HIF-1α/VEGF/VEGFR-2 production using our well-established pig model of CA and resuscitation [7, 9, 23].
Methods
Animal Preparation
All procedures were performed in accordance with institutional guidelines for the care and use of animals established by Capital Medical University (Beijing, China), and the protocol was approved by the Committee on the Ethics of Animal Experiments of Capital Medical University. Twenty-four healthy male domestic Beijing Landrace piglets (12–14 weeks of age, 30.0 ± 2.3 kg) were food restricted overnight and had free access to water. Anesthesia was induced with midazolam (0.5 mg/kg) administered intramuscularly, followed by ear vein injection of propofol (2 mg/kg). Three percent sodium pentobarbital injections (8 mg/kg/h) together with fentanyl (5 μg/kg/h) was given to maintain sedation and analgesia. Pentobarbital was stopped at 18 h after the return of spontaneous circulation (ROSC), followed by intravenous injection of propofol (0.4 mg/kg) when necessary. Before the assessment of neurological outcomes, anesthesia was performed using propofol because recovery from propofol anesthesia is more rapid and complete than recovery from sodium pentobarbital anesthesia. The depth of anesthesia was evaluated based on heart rate, blood pressure, corneal and palpebral reflexes, and bispectral index (Aspect Medical, Newton, MA) [24]. The pigs were intubated and ventilated with a volume-controlled ventilator (Draeger, Evita4, Lubeck, Germany) with a tidal volume of 8 to 15 mL/kg, ventilation rate of 12 to 20 breaths per minute, peak flow of 40 L/min, inspiration-to-expiration ratio of 1:2, and FiO2 of 0.21 to maintain an end-tidal carbon dioxide (ETCO2) between 35 and 45 mmHg. A standard lead II electrocardiogram was used to continuously monitor cardiac rhythm. A central venous catheter was inserted in the right internal jugular vein to measure central venous pressure (CVP) and to allow the administration of fluids and drugs. One 5-F thermistor-tipped arterial catheter was inserted into the femoral artery. The arterial and central venous catheters were connected to the PiCCO system (Pulsion Medical Systems, Munich, Germany) for continuous hemodynamic monitoring, including mean arterial pressure (MAP), heart rate (HR), and discontinuous measurement of cardiac output (CO). Two femoral venous catheters were inserted to place a pacing catheter for the induction of ventricular fibrillation (VF) and to place a central venous cooling catheter for intravascular cooling (Cool Gard XP; Alsius, Los Angeles, CA). One temperature-sensing Foley catheter was intubated into the bladder after fistulation to simultaneously measure the core temperature. All procedures were performed under standard sterile conditions. Room temperature was adjusted to 26˚C.
Experimental Protocol
Baseline measurements were taken 45 min after the completion of the operation in all animals. Four pigs were randomly selected by drawing lots to serve as the sham control group (sham, n = 4) without VF or cooling treatment. The sample size was determined with reference to our previous study and those of other researchers regarding the porcine CA [7, 8, 23]. We calculated that a sample size of seven pigs per group would achieve 90% power to detect a 2.2% decrease in cerebral cortical tissue water content in the HTTM group compared to the NTTM group, with a significance level (alpha) of 0.05. Ultimately, a sample size of ten pigs per group was chosen to allow for a 20% dropout rate.
In 20 of 24 pigs, CA was induced by VF as previously described [7, 9, 23]. VF was electrically induced by programmed electrical stimulation. Mechanical ventilation was discontinued after the onset of VF. After an eight-minute nonintervention interval of VF, basic life support CPR was manually performed at a ratio of 30:2 (compression-to-ventilation) by the same CPR technician. External manual closed chest compression quality was continuously controlled using a HeartStart MRx Monitor/Defibrillator (M3535 A; Philips Medical Systems, Best, Holland) to maintain optimal compressions (rate of 100 ± 5/min and depth of 50 ± 1 mm with complete release). Ventilation was performed using a bag respirator with room air. After 2 min of CPR, defibrillation was attempted by the delivery of a single 120-J biphasic waveform electrical shock with a Smart Biphasic defibrillator (Philips Medical Systems, Andover, MA). If VF persisted, CPR was then resumed for 2 min, and 150 J was used for the second and all subsequent defibrillation attempts every two minutes. ROSC was defined as maintenance of systolic blood pressure > 60 mmHg, which was sustained continuously for at least 10 min. Mechanical ventilation with 100% oxygen was provided after ROSC and continued until 30 min, after which room air was used. Since neurological function recovery is unlikely after 30 min of CA, resuscitation procedures were discontinued if the pigs had no ROSC within 30 min.
Four pigs without ROSC were pronounced dead. Four animals that did not achieve ROSC were not included in the statistical analysis, as the goal was to study the protective mechanism of HTTM on postresuscitation brain injury at 24 h after ROSC. The remaining 16 ROSC animals were randomly divided into two groups: the normothermic targeted temperature management group (NTTM group, n = 8) and the hypothermic (33 °C) targeted temperature management group (HTTM group, n = 8) using the sealed envelope method. Piglets in the HTTM group were immediately cooled after ROSC using the intravascular cooling instrument. The bladder temperature was reduced to 33 °C within 4 h after ROSC [7] and remained at this temperature for 12 h, followed by gradual rewarming (0.5 °C/h) to 37 °C. During induction and maintenance of the target core temperature, the piglets received a continuous infusion of pancuronium (0.2 mg/kg/h) to prevent muscle movement and shivering. The target core temperature of 38 °C was maintained in NTTM and sham piglets using an electric fan and ice bags. The NTTM pigs were treated to the same as the HTTM pigs except for cooling. The resuscitated pigs received intensive care after ROSC. The animals received acetated Ringer’s solution and glucose electrolyte solution to keep MAP above 50 mmHg and CVP above 8 mmHg. If this first step failed, additional norepinephrine was administered to keep MAP above 50 mmHg.
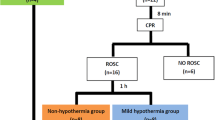
Sedatives and analgesics were discontinued 23 h after ROSC. All vascular catheters were removed, and the abdominal cavity was closed. Animals were allowed to recover from anesthesia, placed in observation cages, and monitored every 15 min until 24 h after ROSC. Neurological outcomes in pigs were assessed by two independent researchers using neurological deficit scores (NDSs) at 24 h after ROSC as described in a previous report [23]. Briefly, the NDS reflects the level of consciousness, motor and sensory function, respiratory pattern, and behavior. The neurological deficits were scored from 0 (no neurological deficit) to 400 (death or brain death). The pigs were then euthanized with an overdose of potassium chloride intravenously. Tissue specimens from the frontal cortex were harvested and immediately frozen in liquid nitrogen and stored at − 80 °C. The experimental procedure is illustrated in Fig. 1.

Experimental procedure. VF ventricular fibrillation, CPR cardiopulmonary resuscitation, ROSC return of spontaneous circulation, NTTM normothermic targeted temperature management, HTTM hypothermic targeted temperature management
Measurement
Assessment of Brain Edema
The brain water content, a marker of brain edema, was determined by measuring the wet-to-dry ratio in piglets at 24 h after ROSC as described in detail in our earlier report [7].
Blood Biochemical Assays
Arterial blood samples were drawn from the femoral artery at baseline and at 0.5, 6, 12, and 24 h after ROSC and centrifuged to measure the protein concentrations of HIF-1α, VEGF, VEGFR-2, and neuron-specific enolase (NSE) with enzyme immunoassay kits (Biosynthesis Biotechnology, Beijing, China). The isolated serum was immediately frozen at − 80 °C and stored until analysis. All assays were carried out in triplicate.
Real-Time Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from brain cortical tissue using TRIzol reagent (Invitrogen Corporation, USA). RT-PCR was performed using the SYBR® Premix Ex Taq™ Kit (Takara, Dalian, China) and the Real-Time PCR Detection System (Bio-Rad, USA). The sequences of primers were as follows: HIF-1α [5′- GAGAAGTCTAGAGATGCAGCCAG-3′ (sense) and 5′ GGTAAGCCTCATAACAGAAGCCT-3′ (antisense)], VEGF [5′-CCTTGCTGCTCTACCTCCAC-3′ (sense) and 5′- ACTCCAGACCTTCGTCGTTG-3′ (antisense)], VEGFR-2 [5′- AACGAGTGGAGGTGACAGATTG-3′ (sense) and 5′- CGGGTAGAAGCACTTGTAGGC-3′ (antisense)] and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [5′-TTGTGATGGGCGTGAA-3′ (sense) and 5′-TCTG GGTGGCAGTGAT-3′ (antisense)]. GAPDH served as an internal control. The relative expression of each target gene compared to the expression of GAPDH was analyzed using the 2−ΔΔCT method.
Western Blotting Analysis
Proteins from cortical tissues were prepared by rapid homogenization in Tissue Extraction Reagent II (Invitrogen Corporation, Carlsbad, CA, USA) according to the manufacturer’s instructions. Lysates were centrifuged (15,000 rpm, 15 min, 4 °C), and their protein concentrations were determined using a BCA kit (Bio-Rad, Hercules, CA, USA). Protein extracts (50 µg) were added to 8%–12% SDS–polyacrylamide gels and subsequently electrotransferred onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA), which were blocked for 2 h with 5% nonfat milk in TBST (Tris-buffered saline with 0.1% Tween-20) prior to immunoblotting overnight at 4 °C with the following primary rabbit antibodies against HIF-1α (1:1000; Biosynthesis Biotechnology, Beijing, China), VEGF (1:1000, Biosynthesis Biotechnology), VEGFR-2 (1:300, Biosynthesis Biotechnology), and β-actin (1:1000, Biosynthesis Biotechnology, Beijing, China). Membranes were incubated with HRP-conjugated secondary antibodies (1:20,000) for 2 h at room temperature. β-actin was used as an endogenous control for total protein, and bands were detected using an enhanced chemical luminescence system (Tanon, Shanghai, China). Integrated optical densities (IODs) of protein bands were digitally quantified using Gel-Pro Analyzer version 3.1 (Media Cybernetics, Silver Spring, MD). Protein expression levels normalized to β-actin are presented as the resulting ratio.
Statistical Analysis
The statistical analysis was performed using IBM SPSS software (version 21.0, Armonk, NY, USA). Continuous variables are presented as the mean ± standard deviation (SD) for normally distributed data. Nonnormally distributed data were expressed as the median and the 25th and 75th percentiles. Student’s t test was used to compare the CPR time before ROSC and the postresuscitation hemodynamic variables between two groups. Baseline values and postresuscitation brain water content, cerebral cortical HIF-1α, VEGF, VEGFR-2 mRNA, and protein expression were compared using one-way analysis of variance (ANOVA) to assess the differences among the three groups, followed by post hoc t tests with a Bonferroni correction to account for multiple comparisons. Changes in HIF-1, VEGF, VEGFR-2, and NSE over time were compared using repeated-measures ANOVA with a Bonferroni post hoc test. The number of shocks before ROSC exhibited a skewed distribution and was analyzed using the Mann–Whitney U test. The Kruskal–Wallis test was performed to identify the overall difference of NDS among the three groups, and the Mann–Whitney U test was further performed to identify the difference between groups. Dichotomous outcomes (24-h survival rate) were compared using the Fisher’s exact test. A two-sided p value < 0.05 was considered statistically significant.
Results
Baseline Status, Resuscitation Outcomes, Survival and Neurological Outcomes
No significant differences were observed in baseline characteristics, including body weight, bladder temperature, and hemodynamics, among the three groups (Table 1). There was no significant difference in the number of shocks [2.0 (1.0, 3.0) vs 2.0 (1.25, 3.75), p = 0.72] or CPR time (4.8 ± 2.5 min vs 5.2 ± 2.4 min, p = 0.71) before ROSC between the NTTM and HTTM groups. Two resuscitated pigs in the NTTM group (25%) did not survive for 24 h, and all resuscitated pigs in the HTTM group survived for 24 h, but the difference in survival between the two groups did not reach statistical significance (p = 0.47). The NDS score was higher (both p < 0.01) in the NTTH group [150.0 (70.0, 177.5), n = 8] and in the HTTH group [237.5 (190.0, 371.3), n = 8] than in the sham group [0 (0, 0), n = 4] 24 h after ROSC. However, the NDS score of the HTTM group was significantly better than that of the NTTM group (p = 0.002).
There were no significant differences in MAP or HR between the NTTM and HTTM groups at 0.5 h [MAP, 89.5 ± 9.0 vs. 90.0 ± 11.3, p = 0.92; HR, 100.0 ± 8.0 vs. 105.4 ± 7.9, p = 0.19], 6 h [MAP, 91.3 ± 8.5 vs. 91.0 ± 7.6, p = 0.95; HR, 111.0 ± 12.0 vs. 109.9 ± 9.8, p = 0.83], 12 h [MAP, 89.0 ± 10.0 vs.87.3 ± 11.3 p = 0.20; HR, 114.7 ± 11.5 vs. 118.4 ± 16.1, p = 0.64], or 24 h [MAP, 90.7 ± 8.2 vs. 89.0 ± 10.3, p = 0.50; HR, 107.8 ± 12.2 vs. 108.1 ± 10.5, p = 0.96] after ROSC. There were no significant differences in CO between the NTTM and HTTM groups at 0.5 h [3.3 ± 0.5 vs. 3.3 ± 0.4, p = 0.77], 6 h [3.1 ± 0.5 vs. 2.9 ± 0.3, p = 0.22], or 24 h [3.8 ± 0.4 vs. 3.5 ± 0.4, p = 0.21] after ROSC. At 12 h after ROSC, CO was significantly lower in the HTTM group than in the NTTM group [3.0 ± 0.4 vs. 3.5 ± 0.4, p = 0.03]. Cumulative crystalloid fluid load and cumulative norepinephrine doses were not significantly different between groups 24 h after ROSC [fluid load (p = 0.60), norepinephrine doses (p = 0.84); NTTM: 4161 ± 650 mL, 4.6 ± 1.3 mg; HTTM: 3957 ± 720 mL, 4.5 ± 1.1 mg].
In the sham and NTTM groups, the core temperature ranged from 37.9 °C to 38.2 °C after ROSC. The core temperature in the HTTM group decreased to the target temperature (33 °C) at the same rate using the intravascular cooling instrument. Cooling to 35 °C required 110.3 ± 4.0 min, to 33 °C required 212.6 ± 5.1 min.
Brain Water Content
Cortical tissue water content was higher in the NTTH group (83.25% ± 1.30%) and in the HTTH group (81.15% ± 1.12%) than in the sham group (78.57% ± 1.49%, both p < 0.05) 24 h after ROSC. However, the cortical tissue water content was lower in the HTTH group than in the NTTM group (p = 0.02).
Serum HIF-1α, VEGF, VEGFR-2, and NSE Levels
Two resuscitated animals in the NTTM group did not survive 24 h. One pig died at 4 h after ROSC, and one pig died at 10 h after ROSC; the cause of death in both cases was refractory shock. The serum HIF-1α, VEGF, VEGFR-2, and NSE values at specific time points were measured and analyzed in both animals before death because we studied the changes in these values after ROSC. The serum HIF-1α, VEGF, VEGFR-2, and NSE values at specific time points are shown in Fig. 2. Serum HIF-1α values were increased significantly at 0.5, 6, 12, and 24 h after ROSC in the NTTM and HTTM groups compared with the sham group (all p < 0.01). However, the serum HIF-1α values decreased significantly at 6, 12, and 24 h after ROSC in the HTTM group compared with the NTTM group (all p < 0.01). The serum VEGF, VEGFR-2, and NSE values were higher at 6, 12, and 24 h after ROSC in the NTTM group than in the sham group (all p < 0.05). The serum VEGF and NSE values were higher at 12 and 24 h after ROSC in the HTTM group than in the sham group (all p < 0.05). Importantly, pigs in the HTTM group had lower serum VEGF and NSE levels at 12 and 24 h after ROSC (all p < 0.05) and lower VEGFR-2 levels at 6, 12, and 24 h after ROSC (all p < 0.05) than those in the NTTM group.

Serum protein concentrations of HIF-1α, VEGF, VEGFR-2, and NSE after ROSC. Serum protein levels of HIF-1α, VEGF, VEGFR-2, and NSE were significantly increased in the NTTM and HTTM groups compared with the sham group within 24 h after ROSC. The serum HIF-1α, VEGF, VEGFR-2, and NSE values decreased significantly in the HTTM group compared with the NTTM group within 24 h after ROSC. Data are presented as the mean ± SD. ROSC return of spontaneous circulation, HIF-1α hypoxia-inducible factor-1α, VEGF vascular endothelial growth factor, VEGFR-2 vascular endothelial growth factor receptor subtype 2, NSE neuron-specific enolase, sham sham control group, NTTM normothermic targeted temperature management group, HTTM hypothermic targeted temperature management group. *p < 0.05; **p < 0.01 versus the sham group; #p < 0.05, ##p < 0.01 versus the NTTM group
HIF-1α, VEGF, VEGFR-2 mRNA, and Protein Expression in the Brain 24 h After ROSC
RT-PCR and Western blotting were performed to determine the effects of HTTM on HIF-1α, VEGF, and VEGFR-2 expression in cortical tissues 24 h after ROSC. As shown in Fig. 3, the mRNA expression levels of HIF-1α (5.43 ± 2.23-fold), VEGF (3.29 ± 1.24-fold), and VEGFR-2 (2.97 ± 0.97-fold) were significantly (all p < 0.01) increased in the NTTM group compared with the sham group 24 h after ROSC. However, compared to the NTTM group, the HTTM group had significantly decreased (all p < 0.01) the mRNA expression levels of these proteins in the cortical tissue 24 h after ROSC (HIF-1α: 2.56 ± 1.15-fold relative to the sham group; VEGF: 1.71 ± 0.60-fold relative to the sham group; VEGFR-2: 1.62 ± 0.56-fold relative to the sham group).

HIF-1α, VEGF, and VEGFR-2 mRNA expression in cortical tissues 24 h after ROSC. The mRNA expression levels of HIF-1α, VEGF, and VEGFR-2 in cortical tissue were significantly increased in the NTTM group compared with the sham group 24 h after ROSC. Compared to the NTTM group, the HTTM group had significantly decreased mRNA expression levels of HIF-1α, VEGF, and VEGFR-2 at 24 h after ROSC. The mRNA levels were quantified via RT-PCR in the sham (n = 4), NTTM (n = 6), and HTTM (n = 8) groups, and the results are presented as the mean fold changes ± standard deviations relative to the sham group. Sham sham control group, NTTM normothermic targeted temperature management group, HTTM hypothermic targeted temperature management group. *p < 0.05; **p < 0.01 versus the sham group; #p < 0.05; ##p < 0.01 versus the NTTM group
As indicated in Fig. 4, 24 h after ROSC, Western blotting analysis showed significant increases in HIF-1α, VEGF, and VEGFR-2 (all p < 0.01) in the frontal cortex of NTTM pigs compared with that of sham pigs. However, HTTM pigs had lower expression of these proteins than did NTTM pigs (all p < 0.05).

HIF-1α, VEGF, and VEGFR-2 protein expression in cortical tissues 24 h after ROSC. The protein expression levels of HIF-1α, VEGF, and VEGFR-2 in cortical tissue were significantly increased in the NTTM group compared with the sham group 24 h after ROSC. Compared to the NTTM group, the HTTM group had significantly decreased protein expression levels of HIF-1α, VEGF, and VEGFR-2 at 24 h after ROSC. The protein concentrations of HIF-1α, VEGF, and VEGFR-2 in the cerebral cortices of the sham (n = 4), NTTM (n = 6), and HTTM (n = 8) groups were measured 24 h after ROSC using Western blots. Sham, sham control group; NTTM, normothermic targeted temperature management group; HTTM, hypothermic targeted temperature management group. *p < 0.05; **p < 0.01 versus the sham group; #p < 0.05; ##p < 0.01 versus the NTTM group
Discussion
The main findings of the present study are as follows. (1) Serum protein levels of HIF-1α, VEGF, and VEGFR-2 were significantly increased within 24 h after ROSC in a swine model of CA and CPR. (2) This upregulation in HIF-1α, VEGF, and VEGFR-2 was significantly attenuated by whole-body HTTM, which was immediately induced after ROSC. (3) HTTM also significantly decreased the brain cortical mRNA and protein expression of HIF-1α, VEGF, and VEGFR-2 24 h after ROSC.
HIF-1α is a well-known transcriptional regulator of cellular responses to hypoxia–ischemia, which induces important gene expression regulators involved in erythropoiesis, angiogenesis, apoptosis modulation, and so on [10]. Two previous studies [25, 26] demonstrated that the expression of HIF-1α was upregulated early in the rat cerebral cortex after transient global ischemia. In addition, the expression of VEGF was significantly increased in rat brain cortical, brainstem and hippocampal tissues 24 and 48 h after CA and resuscitation [27]. In a clinical setting, Xiu et al. [28] reported that serum HIF-1α levels increased in patients soon after acute focal cerebral ischemia and might serve as a marker of the severity of neuronal damage and the degree of recovery in ischemic diseases; the higher level of HIF-1α was, the worse the outcome tended to be. Additionally, serum VEGF concentrations are higher in patients with focal cerebral ischemia than in controls [29, 30], and high serum VEGF levels were associated with unfavorable outcomes [30]. In a study by Omar et al. [31], patients with higher VEGF levels tended to have microcirculatory dysfunction after cardiac arrest. In our present study, we employed a pig model and demonstrated that the ischemia/reperfusion injury induced by CA and resuscitation led to the rapid and significant accumulation of serum HIF-1α as early as 0.5 h after ROSC in the NTTM group, to the accumulation of serum VEGF and VEGFR-2 at 6 h, which persisted for at least 24 h, and to the accumulation of HIF-1α, VEGF, and VEGFR-2 24 h after resuscitation in the brain cortex.
The role of HIF-1α in cerebral ischemia is dual. A previous report showed that neuron-specific knockdown of HIF-1α increases hypoxia–ischemia brain damage and reduces the survival of mice subjected to transient focal cerebral ischemia [32]. In contrast, brain-specific knockdown of HIF-1α attenuates brain ischemic injury in the global brain ischemia model of knockout mice [33]. Furthermore, biphasic expression of HIF-1α was observed first at 4 to 8 h and then 2 to 6 days after cerebral ischemia [32, 34]. Early-phase expression increased apoptosis, but late-phase expression facilitated cell survival, and only early selective inhibition of HIF-1α expression provided a neuroprotective effect after cerebral ischemia [34]. In our present study, HTTM was induced immediately and after ROSC, which significantly decreased serum HIF-1α values as early as 6 h after ROSC and decreased brain cortical expression of HIF-1α 24 h after ROSC in this porcine model, suggesting that the neuroprotective effect of HTTM might involve early inhibition of HIF-1α.
HIF-1α regulates many hypoxic effects through mechanisms dependent and independent of its target genes. HIF-1α is a likely mediator of BBB disruption [35], and HIF-1α has been reported to increase BBB permeability mainly through the disruption of brain microvascular endothelial tight junctions [27]. VEGF is a potent growth factor that plays diverse roles in angiogenesis and vasculogenesis, in addition to carrying out neuroprotective and trophic functions in the central nervous system [36]. Previous evidence [37, 38] has demonstrated that VEGF plays a beneficial role after focal cerebral ischemia, potentially decreasing infarct size and improving neurological outcomes, through either neuroprotection or the induction of angiogenesis. In addition, HIF-1α plays a complex role in cerebral ischemia. Zhang et al. [18] have reported that the effects vary over time. Administration of exogenous VEGF promotes angiogenesis in the late (48 h) stage of brain ischemia, improving neurological recovery, whereas early postischemic (1 h) administration of VEGF exacerbates BBB leakage and the risk of hemorrhagic transformation. VEGF is one of the best-known HIF-1α target genes and is also known as vascular permeability factor because of its ability to induce vascular leaks [39]. Thus, one mechanism of HIF-1α-mediated BBB disruption is VEGF upregulation. VEGF increased BBB permeability by changing the redistribution and downregulating the expression of TJ proteins [21, 40, 41]. It has been reported that inhibition of HIF-1α accumulation and the expression of its downstream target VEGF protected the BBB against ischemia/reperfusion-induced injury [42]. VEGF plays an important role in early BBB disruption and vascular leakage, leading to subsequent edema after ischemic brain injury [11, 17, 18, 27]. Indeed, early administration of VEGF increases BBB permeability and worsens neurological outcome in the ischemic brain [18]. Similarly, inhibition of endogenous VEGF in the acute phase attenuates ischemia-induced BBB permeability and brain edema [42, 43]. In addition, VEGF increases vessel permeability through VEGFR-2 activation [21]. As stated above, the HIF-1α/VEGF/VEGFR-2 pathway is likely associated with BBB disruption and cerebral edema. A study on rats by Shen et al. showed that pretreatment with the HIF-1α inhibitor YC-1 alleviated BBB damage after 2 h of ischemia, accompanied by the significant inhibition of matrix metalloproteinase-2 upregulation and by the downregulation of VEGF expression [44]. In our previous study [7], mild hypothermia attenuated CA- and resuscitation-induced early BBB permeability and brain edema by attenuating tight and adherence junction breakdown, but the precise mechanisms of this phenomenon remain to be elucidated. In the present study on pigs, HTTM was associated with an improved neurological outcome and decreased brain edema, which is consistent with previous studies [7, 9, 23]. Furthermore, we found that the HIF-1α/VEGF/VEGFR-2 system was rapidly activated by ischemia–reperfusion injury after ROSC, which suggested that the activation of HIF-1α likely plays a detrimental role in the enhancement of resuscitated BBB disruption and early brain edema formation via VEGF-VEGFR-2 mechanisms. Interestingly, HTTM may reduce the early expression of HIF-1α, leading to a decrease in the expression of its downstream effector VEGF-VEGFR-2. Therefore, based on the above findings, we suggest that the effect of HTTM on the HIF-1α/VEGF/VEGFR-2 system may be partly associated with the preservation of BBB integrity, reducing the increase in CA-evoked early cerebral edema and improving neurological damage after CA and CPR. To the best of our knowledge, the contribution of HIF-1α/VEGF/VEGFR-2 activation to human brain injury after CA remains largely unknown, and our present study suggests that the modulation of HIF-1α/VEGF/VEGFR-2 axis activity may be a potential therapeutic target to reduce brain damage following CA and CPR in the acute phase.
The NSE protein is mainly present in neuronal and peripheral neuroendocrine cells [45]; however, it is also found in small quantities in serum and cerebrospinal fluid. In the event of neuronal damage and BBB disruption, neuronal cells and the BBB become leaky, leading to an increase in NSE serum levels in the blood [46, 47]. Serum NSE protein is a biomarker of brain injury after CA and CPR [46], and increased serum levels of NSE have been demonstrated as a useful tool for predicting neurological outcomes in survivors of CA patients treated with hypothermia [48]. In our study, the serum NSE levels were markedly increased 6 h after ROSC, whereas HTTM significantly reduced the NSE levels following CPR, which suggested that HTTM might protect against CA- and CPR-induced neuronal cell injury and BBB breakdown.
There were a number of limitations in our study. First, young healthy male swine models of CPR do not fully reflect the situation in human patients with underlying diseases, especially heart diseases. Second, neurons in the hippocampus were most sensitive to global ischemic injury; however, our study focused on the cerebral cortex, as it is known to play a central role in the recovery of function after CA. Third, propofol was used to induce and maintain sedation, which has been reported to have some neuroprotective effects against cerebral ischemic/reperfusion injury [49]. Nevertheless, there were no significant differences observed in the amounts of anesthetics among the three groups. Finally, due to the use of an intravascular cooling treatment, it was not possible to blind the investigators to the animals’ temperature condition throughout the experiment; however, two people who were blinded to the treatment assignments analyzed the serum and tissue samples and carried out all the neurological evaluations.
Conclusion
In conclusion, this study demonstrated that HTTM attenuates postresuscitation cerebral injury in a pig model of VF, which may be, at least in part, attributed to suppression of HIF-1α/VEGF/VEGFR-2 signaling pathway activation.
References
Lemiale V, Dumas F, Mongardon N, et al. Intensive care unit mortality after cardiac arrest: the relative contribution of shock and brain injury in a large cohort. Intensive Care Med. 2013;39(11):1972–80.
Rossetti AO, Rabinstein AA, Oddo M. Neurological prognostication of outcome in patients in coma after cardiac arrest. Lancet Neurol. 2016;15(6):597–609.
Dearden NM. Mechanisms and prevention of secondary brain damage during intensive care. Clin Neuropathol. 1998;17(4):221–8.
Callaway CW, Donnino MW, Fink EL, et al. Part 8: Post-Cardiac Arrest Care: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2015;132(18 Suppl 2):S465–82.
Olai H, Thornéus G, Watson H, et al. Meta-analysis of targeted temperature management in animal models of cardiac arrest. Intensive Care Med Exp. 2020;8(1):3.
Arrich J, Holzer M, Havel C, Müllner M, Herkner H. Hypothermia for neuroprotection in adults after cardiopulmonary resuscitation. Cochrane Database Syst Rev. 2016;2(2):Cd004128.
Li J, Li C, Yuan W, et al. Mild hypothermia alleviates brain oedema and blood-brain barrier disruption by attenuating tight junction and adherens junction breakdown in a swine model of cardiopulmonary resuscitation. PLoS ONE. 2017;12(3):e0174596.
Meybohm P, Gruenewald M, Zacharowski KD, et al. Mild hypothermia alone or in combination with anesthetic post-conditioning reduces expression of inflammatory cytokines in the cerebral cortex of pigs after cardiopulmonary resuscitation. Crit Care. 2010;14(1):R21.
Gong P, Li CS, Hua R, et al. Mild hypothermia attenuates mitochondrial oxidative stress by protecting respiratory enzymes and upregulating MnSOD in a pig model of cardiac arrest. PLoS ONE. 2012;7(4):e35313.
Nagle DG, Zhou YD. Natural product-derived small molecule activators of hypoxia-inducible factor-1 (HIF-1). Curr Pharm Des. 2006;12(21):2673–88.
Engelhardt S, Patkar S, Ogunshola OO. Cell-specific blood-brain barrier regulation in health and disease: a focus on hypoxia. Br J Pharmacol. 2014;171(5):1210–30.
Chen C, Hu Q, Yan J, et al. Early inhibition of HIF-1alpha with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol Dis. 2009;33(3):509–17.
Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146(5):1029–39.
van Bruggen N, Thibodeaux H, Palmer JT, et al. VEGF antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J Clin Invest. 1999;104(11):1613–20.
Dobrogowska DH, Lossinsky AS, Tarnawski M, Vorbrodt AW. Increased blood-brain barrier permeability and endothelial abnormalities induced by vascular endothelial growth factor. J Neurocytol. 1998;27(3):163–73.
Argaw AT, Zhang Y, Snyder BJ, et al. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J Immunol. 2006;177(8):5574–84.
Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125(Pt 11):2549–57.
Zhang ZG, Zhang L, Jiang Q, et al. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106(7):829–38.
Dragoni S, Turowski P. Polarised VEGFA signalling at vascular blood-neural barriers. Int J Mol Sci. 2018;19(5):1378.
Suzuki Y, Nagai N, Umemura K. A review of the mechanisms of blood-brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front Cell Neurosci. 2016;10:2.
Fischer S, Wobben M, Marti HH, Renz D, Schaper W. Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc Res. 2002;63(1):70–80.
Chen W, Jadhav V, Tang J, Zhang JH. HIF-1alpha inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol Dis. 2008;31(3):433–41.
Gong P, Zhao H, Hua R, et al. Mild hypothermia inhibits systemic and cerebral complement activation in a swine model of cardiac arrest. J Cereb Blood Flow Metab. 2015;35(8):1289–95.
Johnson KB, Egan TD, Kern SE, et al. Influence of hemorrhagic shock followed by crystalloid resuscitation on propofol: a pharmacokinetic and pharmacodynamic analysis. Anesthesiology. 2004;101(3):647–59.
Chavez JC, LaManna JC. Activation of hypoxia-inducible factor-1 in the rat cerebral cortex after transient global ischemia: potential role of insulin-like growth factor-1. J Neurosci. 2002;22(20):8922–31.
Jin KL, Mao XO, Nagayama T, Goldsmith PC, Greenberg DA. Induction of vascular endothelial growth factor and hypoxia-inducible factor-1alpha by global ischemia in rat brain. Neuroscience. 2000;99(3):577–85.
Pichiule P, Chávez JC, Xu K, LaManna JC. Vascular endothelial growth factor upregulation in transient global ischemia induced by cardiac arrest and resuscitation in rat brain. Brain Res Mol Brain Res. 1999;74(1–2):83–90.
Xue L, Chen H, Lu K, et al. Clinical significance of changes in serum neuroglobin and HIF-1α concentrations during the early-phase of acute ischemic stroke. J Neurol Sci. 2017;375:52–7.
Dassan P, Keir G, Jäger HR, Brown MM. Value of measuring serum vascular endothelial growth factor levels in diagnosing acute ischemic stroke. Int J Stroke. 2012;7(6):454–9.
Zhang Y, Ma T, Hu H, Wang J, Zhou S. Serum vascular endothelial growth factor as a biomarker for prognosis of minor ischemic stroke. Clin Neurol Neurosurg. 2020;196:106060.
Omar YG, Massey M, Andersen LW, et al. Sublingual microcirculation is impaired in post-cardiac arrest patients. Resuscitation. 2013;84(12):1717–22.
Baranova O, Miranda LF, Pichiule P, et al. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27(23):6320–32.
Helton R, Cui J, Scheel JR, et al. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J Neurosci. 2005;25(16):4099–107.
Yeh SH, Ou LC, Gean PW, Hung JJ, Chang WC. Selective inhibition of early—but not late—expressed HIF-1α is neuroprotective in rats after focal ischemic brain damage. Brain Pathol. 2011;21(3):249–62.
Ogunshola OO, Al-Ahmad A. HIF-1 at the blood-brain barrier: a mediator of permeability? High Alt Med Biol. 2012;13(3):153–61.
Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiol Rev. 2009;89(2):607–48.
Marti HJ, Bernaudin M, Bellail A, et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156(3):965–76.
Stowe AM, Plautz EJ, Eisner-Janowicz I, et al. VEGF protein associates to neurons in remote regions following cortical infarct. J Cereb Blood Flow Metab. 2007;27(1):76–85.
Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol. 1999;237:97–132.
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106(6):1977–82.
Wang W, Dentler WL, Borchardt RT. VEGF increases BMEC monolayer permeability by affecting occludin expression and tight junction assembly. Am J Physiol Heart Circ Physiol. 2001;280(1):H434–40.
Yeh WL, Lu DY, Lin CJ, Liou HC, Fu WM. Inhibition of hypoxia-induced increase of blood-brain barrier permeability by YC-1 through the antagonism of HIF-1alpha accumulation and VEGF expression. Mol Pharmacol. 2007;72(2):440–9.
Kimura R, Nakase H, Tamaki R, Sakaki T. Vascular endothelial growth factor antagonist reduces brain edema formation and venous infarction. Stroke. 2005;36(6):1259–63.
Shen Y, Gu J, Liu Z, et al. Inhibition of HIF-1α reduced blood brain barrier damage by regulating MMP-2 and VEGF during acute cerebral ischemia. Front Cell Neurosci. 2018;12:288.
Marangos PJ, Schmechel DE. Neuron specific enolase, a clinically useful marker for neurons and neuroendocrine cells. Annu Rev Neurosci. 1987;10:269–95.
Rech TH, Vieira SR, Nagel F, Brauner JS, Scalco R. Serum neuron-specific enolase as early predictor of outcome after in-hospital cardiac arrest: a cohort study. Crit Care. 2006;10(5):R133.
Michalak S, Kalinowska-Lyszczarz A. The associations between serum vascular endothelial growth factor, tumor necrosis factor and interleukin 4 with the markers of blood-brain barrier breakdown in patients with paraneoplastic neurological syndromes. J Neural Transm (Vienna). 2019;126(2):149–58.
Vondrakova D, Kruger A, Janotka M, et al. Association of neuron-specific enolase values with outcomes in cardiac arrest survivors is dependent on the time of sample collection. Crit Care. 2017;21(1):172.
Lee JH, Cui HS, Shin SK, et al. Effect of propofol post-treatment on blood-brain barrier integrity and cerebral edema after transient cerebral ischemia in rats. Neurochem Res. 2013;38(11):2276–86.
Acknowledgements
This work was supported by the 2015 Annual Special Cultivation and Development Project for Technology Innovation Base of Beijing Key Laboratory of Cardiopulmonary Cerebral Resuscitation Beijing Municipal Science & Technology Commission (No. Z151100001615056).
Author information
Authors and Affiliations
Contributions
CS-L and JB-L contributed to the study design, method development, data interpretation and writing of the manuscript. JB-L took part in the animal experiment, and drafted the manuscript. JY-W helped draft the manuscript. WY, YZ-Z, JL, ZH-L and JY-W took part in the animal experiment, analyzed experimental data and interpreted the results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
ARRIVE guidelines
This study conformed to ARRIVE guidelines.
Source of support
We received the support of the 2015 Annual Special Cultivation and Development Project for Technology Innovation Base of Beijing Key Laboratory of Cardiopulmonary Cerebral Resuscitation Beijing Municipal Science & Technology Commission (No. Z151100001615056).
Conflict of interest
The authors report no conflicts of interest.
Ethical Approval
This study was conducted in strict accordance with guidelines for the care and use of laboratory animals formulated by the Ministry of Science and Technology of the People’s Republic of China.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, J., Li, C., Yuan, W. et al. Targeted Temperature Management Suppresses Hypoxia-Inducible Factor-1α and Vascular Endothelial Growth Factor Expression in a Pig Model of Cardiac Arrest. Neurocrit Care 35, 379–388 (2021). https://doi.org/10.1007/s12028-020-01166-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-020-01166-0