
Abstract
Background
Increased blood glucose and impaired pressure reactivity (PRx) after traumatic brain injury (TBI) are both known to correlate with unfavorable patient outcome. However, the relationship between these two variables is unknown.
Methods
To test the hypothesis that increased blood glucose leads to increased PRx, we retrospectively analyzed data from 86 traumatic brain injured patients admitted to the Neurocritical Care Unit. Data analyzed included arterial glucose concentration, intracranial pressure (ICP), cerebral perfusion pressure (CPP) and end-tidal CO2. PRx was calculated as the moving correlation coefficient between averaged (10 seconds) arterial blood pressure and ICP. One arterial glucose concentration and one time-aligned PRx value were obtained for each patient, during each day until the fifth day after ictus.
Results
Mean arterial glucose concentrations during the first 5 days since ictus were positively correlated with mean PRx (Pearson correlation coefficient = 0.25, p = 0.02). The correlation was strongest on the first day after injury (Pearson correlation coefficient = 0.47, p = 0.008).
Conclusion
Our preliminary findings indicate that increased blood glucose may impair cerebrovascular reactivity, potentially contributing to a mechanistic link between increased blood glucose and poorer outcome after TBI.
Similar content being viewed by others

Introduction
Trauma to the brain causes a cascade of metabolic, haemodynamic, and cerebrovascular events including impaired brain autoregulation, brain edema, cerebral hypoperfusion, hyperemia and catecholamine release [1, 2]. The catecholamine release not only supports the cardiovascular system, but also causes an adrenergic increase in blood glucose concentration [3].
The increase in blood glucose has been shown to correlate with poorer outcomes following head injury [4] leading to a focus on tight glycaemic control protocols [5]. Thus, elevated glucose has been seen as a potentially modifiable ‘secondary insult’, much like hypotension, hypoxia, and intracranial hypertension. This is an attractive therapeutic target as blood glucose levels can be relatively easily controlled by means of an insulin infusion.
However, mechanisms linking elevated blood glucose to poorer outcomes remain obscure. It has been postulated that increased glucose leads to a hyperglycolytic state, which, if combined with cerebral ischemia, leads to an increase in cerebral anaerobic metabolism [6, 7]. While another study has indicated that increased blood glucose may in fact improve cerebral metabolism [8]. An alternative, and as yet unexplored mechanism, is that increased blood glucose impairs cerebral autoregulation, therefore predisposing to further ischemic and hyperemic insults and poor outcome.
Cerebral autoregulation acts to maintain cerebral blood flow across a range of perfusion pressures thus protecting the brain from both hyperemic and ischemic insults [9]. A descriptor of autoregulation is pressure reactivity (PRx) measured as the correlation between arterial blood pressure (ABP) and intracranial pressure (ICP) [10]. A negative correlation implies active pressure reactivity while a positive correlation implies a ‘pressure passive’, impaired pressure reactivity. Like glucose, PRx has been shown to independently predict outcome after traumatic brain injury (TBI) [10]. The relationship between blood glucose and pressure reactivity has not been investigated.
The aim of this study was to determine if increased blood glucose after TBI has any relationship with pressure reactivity. We hypothesized that hyperglycemia would be associated with impaired pressure reactivity.
Methods
Patients and Methods
The data in this study were gathered during a retrospective analysis of data collected prospectively from 86 head-injured patients admitted to the Addenbrooke’s Hospital Neurocritical Care Unit between January 2010 and December 2012. Consecutive traumatic brain-injured patients with a clinical need for ICP monitoring were included for analysis. Informed consent was obtained from all patients (or their next of kin) for the use of collected data for research purposes. The study was approved by the institutional ethics committee (29 REC 97/291).
Inclusion criteria were: TBI as diagnosed on admission; invasive monitoring of ICP and ABP; at least 12 h of monitoring with a coincident blood test. Cerebrovascular data in the 2 h before and 2 h after arterial blood sampling were used.
Patients were managed according to current TBI guidelines [2]. Patients were sedated, intubated, ventilated, and paralyzed. Interventions were aimed at keeping ICP < 20 mm Hg using a stepwise approach of positioning, sedation, muscle paralysis, moderate hyperventilation, ventriculostomy, osmotic agents, and induced hypothermia. Cerebral perfusion pressure (CPP) was maintained >60–70 mm Hg using intravenous fluids, vasopressors, and inotropes. Glucose management was as per insulin sliding scale, with target blood glucose levels of between 6 and 8 mmol/L. Glasgow outcome scale was assessed at 6 months (1—death, 2—persistent vegetative state, 3—severe disability, 4—moderate disability, 5—good recovery).
Data Acquisition and Analyses
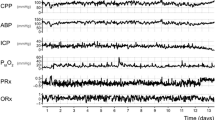
ICP was monitored with an intraparenchymal microsensor inserted into the right frontal cortex (Codman ICP MicroSensor, Codman & Shurtleff, Raynham, MA) with a zero calibration at the level of the foramen of Monro and ABP was monitored in the radial or femoral artery with a zero calibration at the level of the right atrium (Baxter Healthcare CA, USA; Sidcup, UK). End-tidal CO2 data were collected from the ventilator.
Data were sampled at 100 Hz with proprietary data acquisition software (ICM +, Cambridge Enterprise, Cambridge, UK) and stored for subsequent analysis. Data were collected on each day of invasive monitoring until the fifth day after ictus. PRx was calculated as the Pearson correlation of 30 consecutive 10-s average values of ABP and ICP. A 10-s average was used to reduce the influence of respiratory and pulse waveforms. A 300-s moving window was used to generate continuous PRx values. For each day, 4 h of time-averaged cerebrovascular data (CPP, ICP, PRx, end-tidal CO2) were assessed along with one time-aligned arterial blood glucose test taken from the radial or femoral arterial line. Data were analysed only for the first 5 days since ictus.
Statistical Analyses
Ordinal data are presented as medians with their interquartile range (IQR) and continuous data as means and associated standard deviation. Bivariate correlational analyses (Pearson coefficient) between glucose, CPP, ICP, end-tidal CO2, and PRx were calculated. Correlations are zero-order and unadjusted for multiple comparisons. All data analyses were performed on SPSS version 21.0 software (SPSS Inc., Chicago, IL).
Results
Summary details for the 86 patients are shown in Table 1. The median age of the cohort was 41 years with the median initial GCS being 8. 27 % of the patients were female. Mortality at 6 months was 22 %. Mean physiologic monitoring values during the first 5 days since ictus are shown in Table 2. Mean blood glucose was 6.6 mM, PRx was 0.02, ICP was 13.1 mm Hg, CPP was 76.4 mm Hg and PETCO2 was 4.2 kPa. The sample size increased from 31 on the first day since ictus to 60 on the third day since ictus, before falling to 50 on the fifth day since ictus. The varying number of patients at different time points represents the varying time taken between ictus and initiation of intracranial monitoring, and the varying duration of intracranial monitoring.
Mean arterial glucose concentration for each patient during the first 5 days from ictus was significantly correlated with mean PRx (Pearson correlation 0.25, p = 0.02; Table 2; Fig. 1). When just those patients with mean arterial glucose concentration values less than 8 mmol/L were considered, the correlation between glucose concentration and PRx was even stronger (Pearson correlation 0.42, p = 0.0001). Similarly, when paired glucose concentration and PRx values on the just the first day from ictus were considered, this relationship was stronger (Pearson correlation 0.47, p = 0.008, Fig. 2). Pearson correlations between arterial glucose concentration on the second and fifth day since ictus were also statistically significant.

Correlation between mean arterial glucose and PRx in TBI patients during the first 5 days since ictus. There was a significant positive relationship between blood glucose and PRx (Pearson correlation = 0.25; p = 0.02). Each data point represents the mean of the available arterial glucose concentration and PRx measurements during the first 5 days since ictus. If only those subjects with mean arterial glucose concentrations below 8 mmol/L (n = 78) are correlated; the relationship is even stronger (Pearson correlation 0.42; p = 0.0001)

Correlation between arterial glucose concentration and PRx 1 day since ictus. Arterial glucose concentration was positively correlated with PRx (Pearson correlation coefficient 0.47; p = 0.008)
Correlations of potential confounding variables (ICP, end-tidal CO2, CPP) did not approach statistical significance (Table 2).
Discussion
The major finding of this study is a weak but significant positive relationship between arterial glucose concentration and cerebral pressure reactivity in TBI patients. This relationship was strongest when glucose concentration was below 8 mmol/L and when the first day since ictus was considered. We propose that hyperglycemia may lead to poor outcome after TBI, at least in part, due to its hitherto undescribed effect on cerebral pressure reactivity.
Two topics warrant further discussion (1) The relation between hyperglycemia and impaired vascular function, and (2) impaired vascular function as a potential causal link between hyperglycemia and poor outcome after TBI
Effect of Glucose on Vascular Function
In the current study we found a significant correlation between arterial concentration and PRx in TBI patients (Table 2; Fig. 1), indicating that arterial glucose may impair cerebrovascular function.
Limited data of the effect of glucose on vascular function are available, particularly in the cerebral circulation. Some experimental data, however, indicate a glucose-induced impairment of cerebrovascular function. Experimental data from both rats and humans have demonstrated that after administration of glucose, regional cerebral blood flow is decreased [11, 12], while experiments in an isolated pial artery preparation observed that elevated glucose concentrations lead to impaired cerebral endothelial function [13].
In addition to impairing the cerebral circulation, elevated glucose concentration has been shown to impair vascular function systemically. In non-diabetics, oral glucose loading impairs endothelial function as measured with flow-mediated dilation in the upper limb [14] and in healthy humans, oral glucose loading impairs brachial artery myogenic vasoconstriction. [15].
Taken together, these experimental data indicate that elevated blood glucose concentration impairs endothelial and smooth muscle cell responses both in the cerebral and peripheral vasculature. Clearly, further investigation on the link between glucose and pressure reactivity in the TBI population is warranted.
Impaired Vascular Function as a Mediator Between Hyperglycemia and Outcome After TBI
Although not assessed in the current study due to sample size and frequency, the association between elevated glucose and poor outcome after TBI is well-established [4, 16, 17]. However, the physiological significance of this association has been debated. Because hyperglycemia is a common finding in many acute conditions and is related to the initial GCS [4], it is possible that hyperglycemia is epiphenomenal of a systemic stress response and is a reflection of injury severity. Furthermore, recent evidence purports that mild hyperglycemia may in fact be beneficial to cerebral metabolism [18].
Other evidence, however, indicates that after TBI, hyperglycemia per se is harmful. Hyperglycemia leads to an increased anaerobic metabolism and acidosis in the brain [6, 19] which in turn can contribute to neuronal dysfunction and brain edema. Even mild increases in arterial blood glucose are associated with cerebral anaerobic metabolism as assessed by jugular oximetry [20]. In addition, elevated brain glucose concentrations (greater than 5 mM) have been associated with increased concentration of brain glutamate, a substance known for its excitotoxic properties [21].
Our preliminary findings indicate that, in addition to these detrimental metabolic effects, hyperglycemia may impair vascular function. Some support for such a link in disease models can be found in the stroke biology literature. In a rat model, Kawai and colleagues demonstrated that a higher blood glucose leads to a greater size of infarction [22, 23] while in humans, diffusion-weighted MRI in post-stroke patients revealed that increased blood glucose concentration early in the clinical course is associated with an increase in infarction volume [24]. Therefore, evidence indicates that hyperglycemia is associated with a greater ischemic burden, which, combined with our data, leads us to speculate that the adverse effects of hyperglycemia on outcome in TBI could be mediated, in part, by impaired cerebrovascular function.
Limitations
The current study has several important limitations. First, it is possible that the observed relationship could be due to the effect of a confounding physiologic variable (such as ICP, PRx or end-tidal CO2). However, zero-order correlations of these potential confounding factors did not yield any correlations that approached statistical significance and justified inclusion in a multivariate model (Table 2). The influence of other confounders, however, is still possible.
Second, glucose concentration data were collected only daily. Because of this low density of data points, we limited our analysis to the relationship between glucose concentration and PRx between individuals, rather than within individuals. In addition, we have only investigated the relationship between glucose concentration and pressure reactivity early in the disease course (first 5 days since ictus).
Third, glucose concentration was assessed systemically (arterial blood) rather than locally (cerebral microdialysis). Brain tissue glucose concentration could yield additional insight into the relationship between glucose and cerebral autoregulation; however, the focus of this study was to assess the relationship between the prevailing glucose concentration presented to the whole brain, rather than the localized microdialysis glucose concentration in peri-lesional brain tissue.
Finally, although we speculate that a glucose-mediated impairment of cerebral autoregulation may explain, in part, worse outcomes in patients with hyperglycemia, we have not assessed the effect of glucose on outcome in the current study. Such an analysis would likely need a high density of glucose measurements, or glucose measurements very early in the disease course.
Conclusion
We have demonstrated that mild elevation of arterial glucose concentration may impair cerebrovascular reactivity, and propose that this effect could be a mechanistic link between elevated glucose concentration and poor outcome in TBI.
References
Hamill RW, Woolf PD, McDonald JV, Lee LA, Kelly M. Catecholamines predict outcome in traumatic brain injury. Ann Neurol. 1987;21(5):438–43.
Menon DK. Cerebral protection in severe brain injury: physiological determinants of outcome and their optimisation. Br Med Bull. 1999;55(1):226–58.
Yang SY, Zhang S, Wang ML. Clinical significance of admission hyperglycemia and factors related to it in patients with acute severe head injury. Surg Neurol. 1995;3019(95):373–7.
Lam AM, Winn HR, Cullen BF, Sundling N. Hyperglycemia and neurological outcome in patients with head injury. J Neurosurg. 1991;75(4):545–51.
Bilotta F, Caramia R, Cernak I, et al. Intensive insulin therapy after severe traumatic brain injury: a randomized clinical trial. Neurocrit Care. 2008;9(2):159–66.
Rehncrona S, Rosén I, Siesjö BK, et al. Brain lactic acidosis and ischemic cell damage: 1. Biochemistry and neurophysiology. J Cereb Blood Flow Metab. 1981;1:297–311.
Diaz-Parejo P, Ståhl N, Xu W, Reinstrup P, Ungerstedt U, Nordström C-H. Cerebral energy metabolism during transient hyperglycemia in patients with severe brain trauma. Intensive Care Med. 2003;29(4):544–50.
Moro N, Ghavim S, Harris NG, Hovda DA, Sutton RL. Glucose administration after traumatic brain injury improves cerebral metabolism and reduces secondary neuronal injury. Brain Res. 2013;1535:124–36.
Lassen NA. Autoregulation of cerebral blood flow. Circ Res. 1964;15(Suppl):201–4.
Czosnyka M, Smielewski P, Kirkpatrick P, Laing RJ, Menon D, Pickard JD. Continuous assessment of the cerebral vasomotor reactivity in head injury. Neurosurgery. 1997;41:11–7; discussion 17–19.
Duckrow RB, Beard DC, Brennan RW. Regional cerebral blood flow decreases during hyperglycemia. Ann Neurol. 1985;17:267–72.
Page KA, Chan O, Arora J, et al. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA. 2013;309(1):63–70.
Ward ME, Yan L, Angle MR. Modulation of rat pial arteriolar responses to flow by glucose. Anesthesiology. 2002;97(2):471–7.
Title LM, Cummings PM, Giddens K, Nassar BA. Oral glucose loading acutely attenuates endothelium-dependent vasodilation in healthy adults without diabetes: an effect prevented by vitamins C and E. J Am Coll Cardiol. 2000;36(7):2185–91.
Lott MEJ, Hogeman C, Herr M, Gabbay R, Sinoway LI. Effects of an oral glucose tolerance test on the myogenic response in healthy individuals. Am J Physiol Circ Physiol. 2007;292:H304–10.
Van Beek JGM, Mushkudiani NA, Steyerberg EW, et al. Prognostic value of admission laboratory parameters in traumatic brain injury: results from the IMPACT study. J Neurotrauma. 2007;24(2):315–28.
Pentelényi T, Kammerer L. Changes in blood glucose after head injury and its prognostic significance. Injury. 1977;8(4):264–8.
Vespa P, McArthur DL, Stein N, et al. Tight glycemic control increases metabolic distress in traumatic brain injury. Crit Care Med. 2012;40:1923–9.
Zygun DA, Steiner LA, Johnston AJ, et al. Hyperglycemia and brain tissue pH after traumatic brain injury. Neurosurgery. 2004;55:877–81; discussion 882.
Holbein M, Béchir M, Ludwig S, et al. Differential influence of arterial blood glucose on cerebral metabolism following severe traumatic brain injury. Crit Care. 2009;13(1):R13.
Meierhans R, Béchir M, Ludwig S, et al. Brain metabolism is significantly impaired at blood glucose below 6 mM and brain glucose below 1 mM in patients with severe traumatic brain injury. Crit Care. 2010;14(1):R13.
Yip PK, He YY, Hsu CY, Garg N, Marangos P, Hogan EL. Effect of plasma glucose on infarct size in focal cerebral ischemia-reperfusion. Neurology. 1991;41:899–905.
Kawai N, Keep RF, Betz AL. Hyperglycemia and the vascular effects of cerebral ischemia. Stroke. 1997;28(1):149–54.
Baird TA, Parsons MW, Phan T, et al. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke. 2003;34(9):2208–14.
Acknowledgments
We express our thanks to the whole NCCU team, the patients, and their families. JD is supported by a Woolf Fisher scholarship. GVV is supported by an A. G. Leventis Foundation Scholarship and a Charter Studentship from St Edmund’s College, Cambridge. PJH is supported by an NIHR Research Professorship, the NIHR Cambridge Biomedical Research Centre and has been appointed as the Surgical Specialty Lead for Neurosurgery, Royal College of Surgeons of England Clinical Research Initiative. KLHC is supported by the NIHR Biomedical Research Centre, Cambridge (Brain Injury and Repair Theme). MC is supported by NIHR Biomedical Research Centre in Cambridge, UK.
Conflict of interest
ICM+ Software is licensed by Cambridge Enterprise, Cambridge, UK, http://www.neurosurg.cam.ac.uk/icmplus/. MC and PS have a financial interest in a fraction of the licensing fee. The corresponding author and the rest of the co-authors do not have any conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Donnelly, J., Czosnyka, M., Sudhan, N. et al. Increased Blood Glucose is Related to Disturbed Cerebrovascular Pressure Reactivity After Traumatic Brain Injury. Neurocrit Care 22, 20–25 (2015). https://doi.org/10.1007/s12028-014-0042-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12028-014-0042-4