
Abstract
Although fructosyltransferases from Aspergillus aculeatus have received a considerable interest for the prebiotics industry, their amino acid sequences and structural features remain unknown. This study sequenced and characterized a fructosyltransferase from A. aculeatus (AcFT) isolated by heat treatment of Pectinex Ultra SP-L. The AcFT enzyme showed two isoforms, low-glycosylated AcFT1 and high-glycosylated AcFT2 forms, with similar optimum activity at 60 °C. The purified heat-resistant AcFT1 and AcFT2 isoforms produced identical patterns of fructooligosaccharides (FOS; kestose, nystose and fructosylnystose) with a notable transfructosylation capability (~90 % transferase/hydrolase ratio). In contrast, the pI and optimum pH values exhibited discrete differences, attributable to their glycosylation pattern. Partial protein sequencing showed that AcFT enzyme corresponds to Aspac1_37092, a putative 654-residue fructosyltransferase encoded in the genome of A. aculeatus ATCC16872. A homology model of AcFT also revealed the typical fold common to members of the glycoside hydrolase family 32 (GH32), with an N-terminal five-blade β-propeller domain enclosing catalytic residues D60, D191, and E292, linked to a C-terminal β-sandwich domain. To our knowledge, this is the first report describing the amino acid sequence and structural features of a heat-resistant FOS-forming enzyme from A. aculeatus, providing insights into its potential applications in the prebiotics industry.
Similar content being viewed by others

Introduction
The consumption of foods and beverages containing functional prebiotics and probiotics has become a global consumer trend. Particularly, a recent approach has focused on the fortification of beverages with prebiotics such as fructooligosaccharides (FOS) [1, 2]. FOS stimulates the growth of bifidobacteria in the human colon [3] and contributes to a relatively low amount of calories [4], and their consumption decreases the levels of phospholipids, triglycerides, and cholesterol [3, 4]. FOS are mixtures of β-d-fructans, molecules containing between two and four β(2–1)-linked fructosyl units with a terminal α-d-glucose residue; the most representative compounds are 1-kestose (GF2), 1-nystose (GF3), and 1F-fructosylnystose (GF4) [5]. In filamentous fungi and bacteria, these molecules are synthesized by FOS-forming enzymes β-d-fructosyltransferases (FTases; EC 2.4.1.9) or β-fructofuranosidases (FFases; EC 2.4.1.26) through direct transfer of the sucrose fructosyl moiety to an acceptor molecule (transfructosylase activity) that includes sucrose, raffinose, 1-kestose, and nystose [6, 7]. In addition, it has been reported that a few FTases from bacteria unusually transfer the fructosyl moiety to fructosylnystose and fructosylfructosylnystose to produce GF5 and GF6, respectively [8, 9]. Depending on the sucrose concentration and the nature of the hydrophobic residues at the entrance of the active-site pocket where catalytic residues are located, FOS-forming enzymes can also transfer the fructosyl moiety to water providing hydrolase activity and decreasing the synthesis yield of FOS during catalysis [6, 10]. Remarkably, fungal FTases possess almost exclusive transfructosylation activity at high sucrose concentration [10, 11].
According to the carbohydrate-active enzymes database classification (CAZy; http://www.cazy.org/), fungal FOS-forming enzymes belong to the glycoside hydrolase family 32 (GH32). These enzymes are recognized as having molecular weights of ~45–70 kDa, with a typical fold consisting of an N-terminal five-blade β-propeller domain enclosing the negative charged cavity of the active site, linked to a C-terminal β-sandwich domain [12, 13]. Interestingly, fungal FOS-forming enzymes also contain several putative N-linked glycosylation sites that contribute to proper protein folding, thermostability, and function [14]. Specifically, it has been shown that glycosylation is a major determinant of enzymes to thermostability [14–16]. Therefore, fungal FOS-forming enzymes with both heat-resistance and high transfructosylation capability hold great potential for the industrial production of prebiotics [17].
However, the supply of FOS is limited because FOS-forming enzymes are not commercially available. An alternative is to use commercial food-grade enzyme preparations possessing FTase or FFase activity, and accordingly, Pectinex Ultra SP-L, Rohapect CM, Viscozyme L, and Pectinex Smash could serve as a source of food-grade FOS-forming enzymes [18–20]. For instance, Pectinex Ultra SP-L is marketed for use in the processing of fruit juice and contains pectinolytic and cellulolytic enzymes from Aspergillus aculeatus [21, 22]. Nevertheless, Pectinex Ultra SP-L has also been used as the reference preparation because of its known efficient transfructosylating activity [23]. In fact, FTases isolated from commercial enzyme preparations of A. aculeatus showed a high transferase/hydrolase ratio, notable thermal and pH stability, and remarkable activity toward FOS production [19, 24]. However, their amino acid sequences remain unknown, limiting attempts to produce recombinant enzymes and consequently the study of their molecular and structural features.
In this work, we report the amino acid sequence, functional characterization, and structural features of a heat-resistant and glycosylated FTase enzyme (AcFT) isolated from a commercial enzyme preparation of A. aculeatus.
Materials and Methods
Materials and Chemicals
Pectinex ultra SP-L from A. aculeatus (P2611, batch no. 011M2013V) was purchased from Sigma-Aldrich (St. Louis, MO, USA). FOS standards (1-kestose, nystose and 1F-fructofuranosylnystose) were obtained from Wako Pure Chemical Industries (Osaka, Japan). Bradford reagent, acrylamide, bis-acrylamide, Coomassie brilliant blue G-250, molecular weight markers, and immobilized pH gradient (IPG) Ready Strips™ (7 cm, pH 3–10) were purchased from Bio-Rad (Hercules, CA, USA). 3,5-Dinitrosalicylic acid (DNS), sucrose, d-glucose, d-fructose, and all additional reagents were also obtained from Sigma-Aldrich (St. Louis, MO, USA). SDS-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to the method of Laemmli [25] using a Mini-PROTEAN Tetra system (Bio-Rad, Hercules, CA, USA).
Enzyme Purification
Purification of heat-resistant low-glycosylated AcFT1 and high-glycosylated AcFT2 isoforms was performed by three sequential steps: heat treatment, size-exclusion, and anion-exchange chromatography. Specific FTase activity was used as a response variable in the purification process of each step. The crude commercial enzyme preparation Pectinex ultra SP-L was heat-treated over a range of different temperatures (65–80 °C) and time intervals (10–90 min). After heat treatment, non-heat-resistant proteins were removed by centrifugation at 11,000×g for 15 min at 4 °C. Supernatant was then dialyzed against buffer A (50 mM sodium acetate pH 5.5) at 4 °C using dialysis tubing cellulose membrane (12 kDa MWCO, Sigma-Aldrich) under continuous stirring. The dialyzed enzyme was concentrated according to Virgen-Ortíz et al. [26]. The concentrated enzyme solution was then loaded onto a Sephacryl S-200 column (2.5 cm × 55 cm) previously equilibrated with buffer A. The proteins were eluted with the same buffer at a flow rate of 0.5 mL min−1. Fractions with high FTase activity were pooled and applied onto a Q-Sepharose HP chromatographic column (2.5 cm × 10.0 cm) equilibrated with buffer A. Unbound proteins were washed away with several volumes of buffer A. The heat-resistant AcFT1 and AcFT2 isoforms were then eluted with a linear gradient of NaCl (0–0.5 M) in the equilibration buffer at a flow rate of 1.0 mL min−1. The output stream of the column was monitored at 280 nm. The fractions showing FTase activity were dialyzed against buffer A and stored at −80 °C. Protein concentration was estimated by the Bradford dye binding method at 595 nm [27] using bovine serum albumin as a standard in a spectrophotometer Lambda 25 (PerkinElmer, USA).
Optimization Procedure
The optimization methodology used in this work is based on a four-step process: (1) Generating a mesh of experimental data within the design space, (2) modeling the data sets through nonlinear fitting, and (3) using the fitted model to obtain the optimal condition, (4) the optimal calculated value is experimentally confirmed. The dependent variables selected for these studies were the following: X, heat treatment time (min), taking values in the set {10, 20, 30, 45, 60}; Y, temperature (°C), taking values in the set {70, 73, 75, 78, 80}; and Z, the response variable that correspond to the specific activity (U/mg). All the experiments were performed in triplicate.
Enzyme Activity Assay
The global FTase activity was determined by the DNS method [28] following initial rate of the reducing power released from sucrose. One global activity unit (U) is defined as the amount of enzyme that produces 1 μmol of reducing sugar per minute. The assay mixture consisted of 1.0 mL of sucrose (600 g/L) in 50 mM sodium acetate pH 5.5 using 2.5 U mL−1. Reactions were performed at 60 °C in a thermomixer compact (Eppendorf, USA) for 60 min at 300 rpm. The amount of reducing sugar released was calculated using a glucose standard curve prepared under identical conditions.
Effect of pH and Temperature
The activity of AcFT1 and AcFT2 isoforms was determined in the 40–85 °C range using 50 mM sodium acetate pH 5.5. The effect of pH was determined in 50-mM buffer at 60 °C in the 3–8.5 range (citric acid-Na2HPO4 pH 3.0, sodium acetate pH 4.0–5.5, sodium phosphate pH 6.0–8.0, and Tris–HCl pH 8.5).
Analysis of Reaction Mixtures
The transfructosylase and hydrolase ratios (H/T) were determined by measuring the amount of glucose and fructose released in each reaction. The amount of glucose reflects the total amount of sucrose utilized during the reaction (total activity). Fructose is the result of hydrolytic activity, whereas the difference between the glucose and fructose concentration corresponds to the amount of fructose used for FOS synthesis and is therefore the result of the transfructosylation activity. FOS (1-kestose, nystose, and 1F-fructofuranosylnystose) and residual sugars (i.e., sucrose, fructose, and glucose) were analyzed using high-performance liquid chromatography (HPLC). The assay mixture consisted of 1.0 mL of sucrose (400 g/L) in 50 mM sodium acetate pH 5.5 using 2.5 U mL−1. Reactions were performed at 60 °C in a thermomixer compact (Eppendorf, USA) for 0.5, 1, 2, 3, 6, 12, 24, and 48 h at 300 rpm. Samples were manually injected into an HPLC system (Varian Inc., Walnut Creek, CA, USA) equipped with a ternary pump (solvent delivery system model 9012), refractive index detector (model star 9040), and YMC-pack Polyamine II column (4.6 mm × 250 mm) (YMC Inc., Wilmington, NC, USA), kept at 26 °C, using acetonitrile/water (70/30, v/v) as the phase mobile at 1 mL min−1. The residual sugars and FOS were quantified by calibration curves constructed using commercial standards. Unknown FOS were quantified as sucrose, 1-kestose, and nystose, according to their polymerization degree (dihexose, trihexose, and tetrahexose). FOS were also analyzed by HPLC-MS/MS. These analyses were carried using a 6530 Q-TOF mass spectrometer (Agilent Technologies Corp., Palo Alto, CA, USA) equipped with an electrospray ionization source (ESI). The negative ionization mode was applied to acquire MS data, and the capillary voltage was set at 4000 V. High-purity nitrogen (99.5 %) was used as a nebulizing (35 psi) and drying gas (11 L min−1). The gas and vaporizer temperatures were 300 and 350 °C, respectively. The skimmer voltage was set at 65 V, and the fragmentor voltage at 150 V.
Electrophoretic Analysis
Two-dimensional gel electrophoresis (2-DE) with IPG strips was used to determine the isoelectric point (pI) of AcFT1 and its AcFT2 isoforms. IPG strips were passively rehydrated for 16 h at 4 °C in 200 μL of rehydration buffer [8 M urea, 0.05 M dithiothreitol (DTT), 4 % 3-[3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS), 0.2 % ampholytes, and 0.002 % bromophenol blue] containing approximately 100 μg of AcFT1 and AcFT2 isoforms (expressed as total protein). The IPG strip was focused using a Protean IEF cell (Bio-Rad, Hercules, CA, USA) at a constant current of 50 μA per strip under the following conditions: (i) 250 V gradient until 0.01 kVh, (ii) 4000 V gradient until 5.6 kVh, and (iii) holding at 19,000 Vh until 27.3 kVh was reached. After isoelectric focusing, the strips were reduced in an equilibration buffer (6 M urea, 0.05 M Tris–HCl pH 8.8, 2 % SDS, 0.002 % bromophenol blue and 30 % glycerol) containing 1 % DTT over 10 min and then alkylated in the same equilibration buffer containing 2.5 % iodoacetamide for 10 min. For the second dimension, the IPG strip containing the separated proteins was used to load a 12 % polyacrylamide gel with 4 % stacking gel. The pI was estimated according to the protein’s relative location on PAGE.
De Novo Protein Sequencing by LC-MS/MS
The de novo sequencing of AcFT1 and AcFT2 isoforms was performed by Laboratorio Universitario de Proteómica, Instituto de Biotecnología, UNAM (Mexico). Protein spots were manually excised from the 2-DE gels and subjected to MS analysis. Gel pieces were destained, reduced in dithiothreitol, alkylated with iodoacetamide, and digested with trypsin. The sequence of each peptide was analyzed on an Accela micro-flow LC-MS/MS system (Thermo Fisher, Co., San Jose, CA, USA) with a (1/20) splitter and coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo-Fisher Co., San Jose, CA, USA) equipped with a nano-electrospray ionization detector. The equipment was calibrated with a mixture of 10 calibration molecules (Calmix) to allow determination of mass with an accuracy within 5 ppm. Chromatography of peptides prior to mass spectral analysis was accomplished using a micro-capillary column PicoFrit Proteopep 2 C18 (75 μm × 50 mm) (New Objective Inc., Woburn, MA, USA). Separation was achieved by applying a gradient of acetonitrile (10–100 %) in 0.1 % acetic acid in water over a 120-min gradient at a flow rate of 400 nL min−1. Collision-induced dissociation and high-energy collision dissociation methods were used to fragment the peptides. Only 2+- and 3+-charged ions were selected for fragmentation events, while singly charged ions, 4+- or higher-charged ions, and ions for which no charge state could be determined were excluded from selection. All mass spectra were acquired in the positive-ion mode. MS data were acquired using a data-dependent method dynamically choosing the most abundant precursor ions from the survey scan for fragmentation. Mass spectra were collected using normalized collision energy of 35 %, isolation width of 3.0 (m/z), activation Q of 0.25, activation time of 40 ms, and maximum ion injection time of 10 ms per micro-scan. Dynamic exclusion was used to reduce the redundancy of the data. Dynamic exclusion settings were (i) exclusion list size of 500, (ii) pre-exclusion duration of 15 s, and (iii) exclusion duration of 60 s. The MS data set produced by the amino acid sequence of the corresponding peptides was searched against the National Center for Biotechnology Information (NCBI) nonredundant database using Mascot software (Matrix Science, Boston, MA) and Protein Prospector (http://prospector2.ucsf.edu/prospector/cgi-bin/msform.cgi?form=batchtagweb). The principal homology searching protocol for de novo sequences was the MS BLAST web server against nr95 database using the LC–MS/MS Presets option (http://genetics.bwh.harvard.edu/msblast/index.html) and Mascot search results.
Sequence Analysis
Peptide sequences obtained by de novo sequencing were analyzed using the Joint Genome Institute database (JGI/MycoCosm) (http://jgi.doe.gov/fungi) [29, 30] and BLAST search algorithm (http://blast.ncbi.nlm.nih.gov/). All peptides of both AcFT1 and AcFT2 isoforms were compared against the genome of A. aculeatus ATCC16872 (JGI Project Id: 52394, http://genome.jgi-psf.org/pages/search-for-genes.jsf?organism=Aspac1), including all proteins before and after filtering. The analysis unambiguously selected a single protein encoded in the genome of A. aculeatus ATCC16872, a 654-residue hypothetical fructosyltransferase annotated as Aspac1_37092 (Protein Id: 1877157). Sequence analysis of Aspac1_37092 enzyme was then performed using nonredundant protein-protein BLAST algorithm and Pfam (http://pfam.xfam.org/) [31]. The putative signal peptide cleavage site was predicted with the SignalP 4.1 server (http://www.cbs.dtu.dk/services/SignalP/). The theoretical isoelectric point and molecular weight were calculated using the Expasy Compute pI/Mw tool (http://web.expasy.org/compute_pi/). Sequence alignments were performed using CLUSTAL W with default settings [32].
Glycosylation Analysis
The glycosylation in AcFT1 and its AcFT2 isoforms was tested using a GelCode glycoprotein staining kit (Pierce, Rockford, IL, USA) according to the manufacturer’s instructions. Horseradish peroxidase (~45 kDa) was used as a positive control and soybean trypsin (~27 kDa) as a negative control. The 12 % SDS-PAGE was placed in periodic acid solution (mixed 3 % acetic acid) for 60 min at room temperature and then soaked with distilled water for 60 min. After staining with Schiff’s reagent for 1 h, the gel was decolorized with 1 % sodium sulfite.
N-Glycosylation Site Prediction and Modeling
The prediction of the AcFT enzyme N-glycosylation sites was performed using NetNGlyc 1.0 (http://www.cbs.dtu.dk/services/NetNGlyc/) and NGlycPred Server [33]. To analyze the role of N-glycosylation sites in AcFT structure, a homology model was generated using the CPHmodels 3.2 server [34]. The modeled three-dimensional structure was built on the basis of sequence identity with the crystallographic structure of fructosyltransferase from Aspergillus japonicus CB05 (AjFT; PDB entry 3LF7) [13] which shares 96 % sequence identity with AcFT enzyme. The model was subjected to energy minimization using YASARA software [35] and then validated using MolProbity [36]. Structural analysis was performed using Coot [37] and the PDBsum tool [38]. The graphical representations were made using CCP4mg version 2.10.4 [39].
Statistical Analysis
All experiments were performed in triplicate and expressed as mean ± standard deviations, unless otherwise stated. Statistical significance was assessed with one-way analysis of variance (ANOVA) using the MatLab software (MathWorks, USA). Treatment means were considered significantly different at P ˂ 0.05.
Results and Discussion
Purification of Heat-Resistant AcFT1 and AcFT2
Since FTases from A. aculeatus have relative heat-resistance [19, 24], a heat treatment procedure was applied to selectively purify heat-resistant FTases from Pectinex Ultra SP-L, a commercial enzyme preparation which contains a mixture of enzymes from A. aculeatus. Accordingly, the enzyme preparation Pectinex Ultra SP-L was treated over a range of different temperatures (65–80 °C) and time intervals (10–90 min) (Fig. 1a, b). As illustrated in the 3D surface plot and 2D contour plot (Fig. 1a), maximal specific FTase activity was obtained using a heat treatment of 75 °C for 46 min, where the specific activity of FTase increased 8.2-fold (Table 1). Since nonthermostable proteins are denatured when the temperature is increased [40], the optimum heat treatment (46 min, 75 °C) allowed the semi-purification of heat-resistant FTases, removing about ~90 % of the contaminating proteins as compared to the crude extract (Fig. 1b, lane 4). The same strategy has been reported to selectively purify thermostable cellulases [41] and β-glycosidases [42] from crude extracts.

Purification of heat-resistant AcFT1 and AcFT2. a 3D surface plot and 2D contour plot showing the effect of temperature and time on the FTase activity during the heat treatment applied in the first partial purification step. b Coomassie-stained SDS-PAGE profiles after heat treatment. Protein molecular weight marker (lane M), crude extract (lane 1), 70 °C for 10 min (lane 2), 75 °C for 10 min (lane 3), 75 °C for 46 min (optimum) (lane 4), 75 °C for 60 min (lane 5), and 80 °C for 30 min (lane 6)
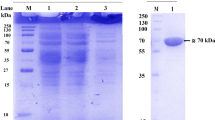
After heat treatment, AcFT1 and AcFT2 were purified to homogeneity by size-exclusion and anion-exchange chromatography. AcFT1 and AcFT2 showed a molecular mass of ~70 and ~100 kDa, respectively, on SDS-PAGE (Fig. 2a). Notably, glycoprotein staining revealed that both AcFT1 and AcFT2 were glycosylated (Fig. 2b). In accordance with these results, it has been shown that FOS-forming enzymes isolated from crude extracts of Aspergillus spp. have a carbohydrate content of 1–30 % [14, 24, 43]. In fact, heat-resistant and glycosylated FTases with a similar molecular mass to AcFT1 and AcFT2 have also been isolated from A. aculeatus, A. japonicus, and A. niger [7, 24, 44]. The results of the purification steps are summarized in Table 1.

Relative electrophoretic mobility on SDS-PAGE of AcFT1 (~70 kDa) and AcFT2 (~100 kDa). a SDS-PAGE analysis after anion-exchange chromatography. Protein molecular weight marker (lane M), AcFT2 (lane 1), and AcFT1 (lane 2). b Glycoprotein staining. AcFT1 (lane 1) and AcFT2 (2 μg); nonglycosylated soybean trypsin inhibitor (~27 kDa) (lane 2); glycosylated horseradish peroxidase (~45 kDa) (lane 3). c 2-DE gel analysis to determine the isoelectric point (pI)
Sequence Analysis
In order to identify heat-resistant FTases from A. aculeatus (Fig. 2), the AcFT1 and AcFT2 internal amino acid sequences were obtained by de novo sequencing using in-gel digestion with trypsin and LC-MS/MS analysis. Thus, the amino acid sequences of eight peptides for AcFT1 and six peptides for AcFT2 were determined, providing a total of 121 and 87 residues, respectively. Interestingly, all AcFT2 peptides sequences were identical to AcFT1 (Table 2), indicating that both glycoproteins are isoforms of the same enzyme. The BLAST alignment of all peptides revealed 99.7 % sequence identity to a single protein Aspac1_37092, encoded in the genome of A. aculeatus ATCC16872, with only one exception, the proline of the AcFT1 SSPLQSGFEILASELER peptide and AcFT2 isoforms annotated as asparagine in the genome of A. aculeatus ATCC16872 (Fig. 3). Sequence analysis also showed that the Aspac1_37092 coding sequence of 1965 base pairs corresponds to a putative 654-residue fructosyltransferase, with an N-terminal amino acid sequence (LDTTAPPPTNLSTLPNNTLF) similar to the partial N-terminal amino acid sequence (LDTTAPPXXFLSTLPXXXLF) of a heat-resistant and glycosylated FTase enzyme also isolated from the commercial enzyme preparation Pectinex Ultra SP-L of A. aculeatus (Fig. 3) [16]. The Aspac1_37092 full-length protein (hereinafter referred as AcFT) contained a signal peptide of 20 residues (MKLPTTTLALATGAAAAAA) which shares 90 % sequence identity with previously reported signal peptides FTases and FFases from Aspergillus spp. (Fig. 3). In addition, the 654-residue AcFT enzyme showed similar domains common to members of the GH32 family and a theoretical molecular weight of 70.6 kDa. Members of GH32 family are widely recognized for their β-fructofuranosidase or fructosyltransferase activities in filamentous fungi [12]. Notably, AcFT enzyme shares 96 % sequence identity with AjFT enzyme (NCBI accession no. ADK46938.1) and β-fructofuranosidase FopA from Aspergillus niger (NCBI accession no. BAB67771.1) (Fig. 3). The mature form of AcFT enzyme presents a theoretical pI and molecular weight values of 4.6 and 68.9 kDa, respectively. This molecular weight is consistent with the AcFT1 isoform on SDS-PAGE (~70 kDa) (Fig. 2). However, the molecular weight of ~100 kDa for the AcFT2 isoform (Fig. 2) is ~30 kDa higher than the theoretical molecular weight of the mature form, suggesting that AcFT1 and AcFT2 glycoproteins are low- and high-glycosylated isoforms. Similar to AcFT, both AjFT and FopA enzymes have been isolated as glycosylated isoforms with molecular weights of ~70–120 kDa [13, 44, 45]. In addition, AcFT1 and its AcFT2 isoforms showed a pI of ~4.5 and ~4.6, respectively, on a 2-DE gel (Fig. 2c), suggesting that the discrete differences in the pI and molecular mass values of both isoforms are attributable to their glycosylation pattern.

Multiple sequence alignment of putative Aspac1_37092 fructosyltransferase from A. aculeatus ATCC16872 with AjFT and FopA enzymes. The amino-acid sequences of all AcFT1 and AcFT2 unique peptides are indicated in the black box. The signal peptide is underlined, and the N-terminal sequence is indicated in gray box. Note that a Pro residue instead of an Asn residue was found at position 522 in both unique peptides of AcFT1 and AcFT2 isoforms
Additionally, a putative 603-residue β-fructofuranosidase enzyme, annotated as Aspac1_54078 was also found in the genome of A. aculeatus ATCC16872 (JGI Project Id: 52394; Protein Id: 54078). Sequence analysis revealed that the Aspac1_54078 enzyme also belongs to the GH32 family and showed theoretical pI and molecular weight values of 5.8 and 67.2 kDa, respectively. However, Aspac1_54078 only shares 23 % sequence identity with the AcFT enzyme and no matches amongst the AcFT1 peptides, the AcFT2 isoforms, and the putative β-fructofuranosidase or other proteins encoded in the genome of A. aculeatus ATCC16872. These results confirm that low-glycosylated AcFT1 and high-glycosylated AcFT2 are isoforms of the putative Aspac1_37092 fructosyltransferase from A. aculeatus ATCC16872. Moreover, AcFT1 is homologous to FTases from Aspergillus spp. (AjFT and FopA), which are known for their high transferase/hydrolase ratio, notable thermal stability, and remarkable activity toward FOS production [43–45].
Effect of pH and Temperature
The optimum temperature of low-glycosylated AcFT1 and high-glycosylated AcFT2 isoforms was 60 °C, indicating that differences in their glycosylation pattern have no effect on the optimal temperature of activity (Fig. 4a). In contrast, the glycosylation pattern of AcFT1 and AcFT2 isoforms affected the optimum pH, where values of 5.5 and 5.0, respectively, were obtained (Fig. 4b). These results are in agreement with those of optimum temperature and pH values determined for fructosyltransferases AjFT [44], FopA [43], and FTase from A. aculeatus [24]. Heat-resistance and high optimum temperature of AcFT1 and AcFT2 isoforms, together with a wide range of pH activity, are advantageous for biotechnological applications, since they reduced the risk of microbial contamination, lower viscosity, and improved transfer rates and solubility of substrates [46].

Effect of temperature (a) and pH (b) on enzyme activity of AcFT1 (empty circles) and AcFT2 (solid circles) isoforms. The error bars indicate the standard deviation of three replicates
Functional Characterization
The transfructosylation capability of AcFT1 and its AcFT2 isoforms toward FOS production was evaluated using 400 g/L of sucrose in 50 mM sodium acetate pH 5.5 with 2.5 U/mL at 60 °C. The main carbohydrates in the reaction mixtures were identified using HPLC by comparing the retention times (t R ) with those of available commercial standard sugars. After a 2-h reaction, HPLC analysis for FOS synthesis showed that low-glycosylated AcFT1 and high-glycosylated AcFT2 isoforms were able to transfer fructosyl motifs from sucrose to produce kestose, nystose, and 1F-fructofuranosylnystose, along with glucose, fructose, and residual sucrose (Fig. 5a). As expected, both isoforms produced identical patterns of end products (Fig. 5a, b). In accordance with these results, the nonglycosylated AjFT enzyme expressed in E. coli showed a similar transfructosylation activity and specificity as the glycosylated enzyme [13].

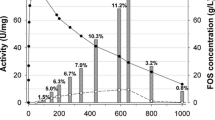
Profile of end products. a HPLC profile of the synthesized FOS from sucrose after 2 h of reaction. b After 24 h of reaction time. Peaks: fructose (1), glucose (2), sucrose (3), 1-kestose (6), nystose (9), 1F-fructosylnystose (11), and unknown (4, 5, 7, 8, 10). c The transfructosylase and hydrolase activity: difference between glucose and fructose (transfructosylation activity), in red; residual fructose (hydrolytic activity), in blue; and FOS formation (1-kestose, nystose, 1F-fructosylnystose), in green, gray, and brown, respectively, while sucrose is in black (color figure online)
The details of the production of the FOS are shown in Fig. 5c. The maximum yield of FOS (313.1 ± 11.8 g L−1) was reached in 2 h. Assuming sucrose conversion as the ratio of transformed sucrose in the reaction relative to the initial sucrose, the total sucrose conversion was 89.7 ± 0.5 %. At 2 h of reaction time, the amount of 1-kestose, nystose and 1F-fructofuranosylnystose was 200.9 ± 5.4, 107.8 ± 5.6, and 4.4 ± 0.8 g L−1, respectively, which give a maximum FOS production of 61.4 ± 1.1 % (w/w) of the total carbohydrates in the reaction mixture. The rest of the carbohydrates in the mixture were fructose (0.7 ± 0.1 %, w/w), glucose (26.4 ± 0.3 %, w/w), and remaining sucrose (11.5 ± 0.8 %, w/w). These values are similar to those reported using the crude Pectinex Ultra SP-L under similar experimental conditions but over a reaction time of 144 h (Ghazi et al. 2005). In other work by Ghazi et al. (2007), similar results were reported about the production of FOS (60.7 %) using pure fructosyltransferase from A. aculeatus for a reaction time of 24 h.
After 24 h, we detected 11 end products (Fig. 5b). The chromatographic behavior and the MS/MS spectra of fructose, glucose, sucrose, 1-kestose, nystose, and 1F-fructofuranosylnystose (peaks 1, 2, 3, 6, 9, and 11, respectively; Table 3) in the samples were similar to those obtained with reference compounds. In addition, five unknown oligosaccharides (peaks 4, 5, 7, 8, and 10; Table 3) were observed at low concentrations in the samples. The content of these compounds was significantly increased after 48 h (Table 3). All of them formed adducts with Cl ([M + Cl]−) and their MS/MS spectra contained ions that reveled the sequential binding of hexoses (m/z of 179.0, 341.1, 503.1, and 665.2) (Table 3). However, the HPLC analysis did not allow the discrimination of glucose and fructose residues in these compounds. Lately, a similar mixture of 1F-FOS, 6F-FOS and 6G-FOS, including the presence of a nonconventional disaccharide, was synthesized by mycelium-bound transfructosylation activity in Cladosporium cladosporioides [47]. According to Zambelli et al. [47], the formation of a mixture of FOS with different glycosidic linkages could give rise to certain benefits regarding their bioactivity. Finally, the transfructosylation capability of AcFT1 and its AcFT2 isoforms, evaluated as the transferase/hydrolase ratio (H/T), revealed that at a sucrose concentration of 400 g/L, both isoforms were able to transfer ~90 % of the fructosyl units toward FOS synthesis (Fig. 5c), demonstrating their high transfructosylation capability.
Homology Modeling
For structural analysis, a three-dimensional structural model of AcFT enzyme was generated by homology modeling using the crystallographic structure of AjFT [13] as a template (Fig. 6). The overall architecture of AcFT is similar to AjFT; a calculated rms deviation of 0.16 Å was obtained after superimposing 633 Cα atoms. The homology model of AcFT comprises the mature form of the enzyme, a 634-residue protein without the first 20 residues of the signal peptide (Fig. 3). The overall fold of AcFT comprises two domains, an N-terminal five-blade β-propeller (residues 21–468) domain, and a C-terminal β-sandwich (residues 478–654) domain, which are linked by a 9-residue short α-helix (residues 469–477). The five-blade β-propeller domain contains five repeats of radially oriented blades (I–V), enclosing a deep central cavity where catalytic residues D60, D191, and E292 are located. The structure alignment of AcFT using the DALI server [48] showed that the enzyme shares a similar fold with enzymes of the GH32 family, revealing structural similarities with invertase from Arabidopsis thaliana (PDB entry 2XQR; DALI Z-score: 36.6; r.m.s.d.: 2.5 Å; sequence identity: 19 %) [49], β-fructofuranosidase from Bifidobacterium longum KN29.1 (PDB entry 3PIJ; DALI Z-score: 36.0; r.m.s.d.: 2.6 Å; sequence identity: 20 %) [50], endo-inulinase from Aspergillus ficuum (PDB entry 3SC7; DALI Z-score: 34.8; r.m.s.d.: 2.8 Å; sequence identity: 18 %) [51], and as expected AjFT (PDB entry 3LF7; DALI Z-score: 71.3; r.m.s.d.: 0.16 Å; sequence identity: 96 %) [13]. Notably, structural comparison of the crystallographic structure of the AjFT enzyme and the AcFT structural model showed that the catalytic residues D60, D191, and E292 (that govern the binding of the terminal fructose and the catalytic reaction at the −1 subsite as well as acting as nucleophile, transition-state stabilizer, and general acid/base catalyst, respectively, along with residues for +1 subsite involved in the binding of fructosyl or glucosyl moieties, +2 subsite for raffinose and +3 subsite for inulin-type substrates with higher structural flexibilities) are all identical in both enzymes. Together, these results confirm that the AcFT enzyme belongs to the GH32 family and its catalytic properties are associated with the synthesis of FOS (Fig. 5).

Homology model of the AcFT enzyme. The three-dimensional structure model consists of an N-terminal five-blade β-propeller (I-V, residues 21–468, dark cyan) domain, a C-terminal β-sandwich (residues 478–654, lawn green) domain, and a 9-residue short α-helix (residues 469–477, yellow) that connects both domains. Note that the five blades (I–V) enclose a central cavity with catalytic residues D60, D191, and E292 (black cylinders). The asparagines predicted to be N-glycosylated are given red ball-and-stick representation (color figure online)
As mentioned above, glycosylation is one of the naturally occurring covalent modifications of the eukaryotic proteins in Aspergillus spp., which commonly present a carbohydrate content that represents 1–80 % of the total weight [52]. Accordingly, 12 potential N-glycosylation sites were predicted for the AcFT enzyme occurring as asparagines located in the NX(S/T) motif; these include N32, N38, N101, N128, N213, N255, N274, N455, N478, N545, N621, and N639. All 12 glycosylation sites were exposed at the surface of AcFT structure and are distant from the catalytic site formed by D60, D191, and E292 in the central cavity (Fig. 6). It has previously been shown that glycans commonly contribute to the proper folding of the enzymes, as well as increased solubility and stability against proteolysis and thermostability [53]. In addition, it has also been shown that the degree of enzyme thermostability is mainly dependent on the position of the glycosylation sites but very weakly on the size of the glycans [53]. In contrast, the total absence of N-linked oligosaccharides in the nonglycosylated E. coli-expressed β-fructosidase from Aspergillus oryzae FS4 (BfrA) resulted in a significant decrease in thermostability, compared to the N-glycosylated native and the P. pastoris-expressed enzyme [14]. Therefore, since the position of the 12 potential N-glycosylation sites of heat-resistant isoforms AcFT1 and AcFT2 is the same and they showed a similar optimum activity at 60 °C and similar patterns of FOS synthesis, these results suggest that differences in their isoelectric point, optimum pH, and molecular weight values are owing to the size of glycans of both isoforms.
Conclusions
In this study, we report for the first time the amino acid sequence and structural features of a heat-resistant FOS-forming enzyme (AcFT) from A. aculeatus, which showed a high transferase/hydrolase ratio. Combined with its functional properties toward FOS production, the AcFT enzyme holds great potential for the prebiotics industry.
References
Renuka, B., Kulkarni, S. G., Vijayanand, P., & Prapulla, S. G. (2009). Fructooligosaccharide fortification of selected fruit juice beverages: effect on the quality characteristics. LWT - Food Science and Technology, 42(5), 1031–1033.
Flores-Maltos, D. a., Mussatto, S. I., Contreras-Esquivel, J. C., Rodríguez-Herrera, R., Teixeira, J. a., & Aguilar, C. N. (2015). Biotechnological production and application of fructooligosaccharides. Critical Reviews in Biotechnology, 8, 1–9.
Roberfroid, M. B. (2007). Inulin-type fructans: functional food ingredients. The Journal of Nutrition, 137, 2493–2502.
Mussatto, S. I., & Mancilha, I. M. (2007). Non-digestible oligosaccharides: a review. Carbohydrate Polymers, 68(3), 587–597.
Crittenden, R. G., & Playne, M. J. (1996). Production, properties and applications of food-grade oligosaccharides. Trends in Food Science and Technology, 7(11), 353–361.
Antosova, M., & Polakovic, M. (2001). Fructosyltransferases: the enzymes catalyzing production. Chemical Papers, 55(6), 350–358.
Hirayama, M., Sumi, N., & Hidaka, H. (1989). Purification and properties of a fructoooligosaccharide-producing beta-fructofuranosidase from Aspergillus niger ATCC 20611. Agricultural and Biological Chemistry, 53(3), 667–673.
Park, J. P., Oh, T. K., & Yun, J. W. (2001). Purification and characterization of a novel transfructosylating enzyme from Bacillus macerans EG-6. Process Biochemistry, 37(5), 471–476.
Díez-Municio, M., de las Rivas, B., Jimeno, M. L., Muñoz, R., Moreno, F. J., & Herrero, M. (2013). Enzymatic synthesis and characterization of fructooligosaccharides and novel maltosylfructosides by inulosucrase from Lactobacillus gasseri DSM 20604. Applied and Environmental Microbiology, 79(13), 4129–4140.
Ganaie, M. A., Gupta, U. S., & Kango, N. (2013). Screening of biocatalysts for transformation of sucrose to fructooligosaccharides. Journal of Molecular Catalysis B: Enzymatic, 97, 12–17.
Guio, F., Rugeles, L. D., Rojas, S. E., Palomino, M. P., Camargo, M. C., & Sánchez, O. F. (2012). Kinetic modeling of fructooligosaccharide production using Aspergillus oryzae N74. Applied Biochemistry and Biotechnology, 167(1), 142–163.
Lammens, W., Le Roy, K., Schroeven, L., Van Laere, A., Rabijns, A., & Van Den Ende, W. (2009). Structural insights into glycoside hydrolase family 32 and 68 enzymes: functional implications. Journal of Experimental Botany, 60(3), 727–740.
Chuankhayan, P., Hsieh, C. Y., Huang, Y. C., Hsieh, Y. Y., Guan, H. H., Hsieh, Y. C., & Chen, C. J. (2010). Crystal structures of Aspergillus japonicus fructosyltransferase complex with donor/acceptor substrates reveal complete subsites in the active site for catalysis. Journal of Biological Chemistry, 285(30), 23251–23264.
Xu, L., Wang, D., Lu, L., Jin, L., Liu, J., Song, D., & Xiao, M. (2014). Purification, cloning, characterization, and N-glycosylation analysis of a novel β-fructosidase from Aspergillus oryzae FS4 synthesizing levan- and neolevan-type fructooligosaccharides. PLoS ONE, 9(12), e114793.
Haraguchi, M., Yamashiro, S., Furukawa, K., Takamiya, K., Shiku, H., & Furukawa, K. (1995). The effects of the site-directed removal of N-glycosylation sites from beta-1,4-N-acetylgalactosaminyltransferase on its function. The Biochemical Journal, 312(Pt1 (1)), 273–280.
Clark, S. E., Muslin, E. H., & Henson, C. a. (2004). Effect of adding and removing N-glycosylation recognition sites on the thermostability of barley β-glucosidase. Protein Engineering Design and Selection, 17(3), 245–249.
Maiorano, A. E., Piccoli, R. M., Da Silva, E. S., & De Andrade Rodrigues, M. F. (2008). Microbial production of fructosyltransferases for synthesis of pre-biotics. Biotechnology Letters, 30(11), 1867–1877.
Muñoz-Gutiérrez, I., Rodríguez-Alegría, M. E., & López Munguía, A. (2009). Kinetic behaviour and specificity of β-fructosidases in the hydrolysis of plant and microbial fructans. Process Biochemistry, 44(8), 891–898.
Lorenzoni, A. S. G., Aydos, L. F., Klein, M. P., Rodrigues, R. C., & Hertz, P. F. (2014). Fructooligosaccharides synthesis by highly stable immobilized β-fructofuranosidase from Aspergillus aculeatus. Carbohydrate Polymers, 103(1), 193–197.
Ghazi, I., Gómez De Segura, A., Fernández-Arrojo, L., Alcalde, M., Yates, M., Rojas-Cervantes, M. L., & Ballesteros, A. (2005). Immobilisation of fructosyltransferase from Aspergillus aculeatus on epoxy-activated Sepabeads EC for the synthesis of fructo-oligosaccharides. Journal of Molecular Catalysis B: Enzymatic, 35(1–3), 19–27.
Adisa, V. a., & Fajola, A. O. (1983). Cellulolytic enzymes associated with the fruit rots of Citrus sinensis caused by Aspergillus aculeatus and Botryodiplodia theobromae. Mycopathologia, 82(1), 23–27.
De Vries, R. P., & Visser, J. (2001). Aspergillus enzymes involved in degradation of plant cell wall polysaccharides. Microbiology and Molecular Biology Reviews, 65(4), 497–522.
Vega-Paulino, R. J., & Zúniga-Hansen, M. E. (2012). Potential application of commercial enzyme preparations for industrial production of short-chain fructooligosaccharides. Journal of Molecular Catalysis B: Enzymatic, 76, 44–51.
Ghazi, I., Fernandez-Arrojo, L., Garcia-Arellano, H., Ferrer, M., Ballesteros, A., & Plou, F. J. (2007). Purification and kinetic characterization of a fructosyltransferase from Aspergillus aculeatus. Journal of Biotechnology, 128(1), 204–211.
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 227(5259), 680–685.
Virgen-Ortíz, J. J., Ibarra-Junquera, V., Osuna-Castro, J. a., Escalante-Minakata, P., Mancilla-Margalli, N. a., & Ornelas-Paz, J. D. J. (2012). Method to concentrate protein solutions based on dialysis-freezing- centrifugation: enzyme applications. Analytical Biochemistry, 426(1), 4–12.
Bradford, M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry, 72, 248–254.
Miller, G. L. (1959). Use of dinitrosaiicyiic acid reagent for determination of reducing sugar. Analytical Chemistry, 31(3), 426–428.
Nordberg, H., Cantor, M., Dusheyko, S., Hua, S., Poliakov, A., Shabalov, I., & Dubchak, I. (2014). The genome portal of the Department of Energy Joint Genome Institute: 2014 updates. Nucleic Acids Research, 42(D1), 26–31.
Grigoriev, I. V., Nikitin, R., Haridas, S., Kuo, A., Ohm, R., Otillar, R., & Shabalov, I. (2014). MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Research, 42(D1), 699–704.
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., & Punta, M. (2014). Pfam: the protein families database. Nucleic Acids Research, 42(D1), 222–230.
Larkin, M. a., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. a., McWilliam, H., & Higgins, D. G. (2007). Clustal W and Clustal X version 2.0. Bioinformatics, 23(21), 2947–2948.
Chuang, G. Y., Boyington, J. C., Gordon Joyce, M., Zhu, J., Nabel, G. J., Kwong, P. D., & Georgiev, I. (2012). Computational prediction of N-linked glycosylation incorporating structural properties and patterns. Bioinformatics, 28(17), 2249–2255.
Nielsen, M., Lundegaard, C., Lund, O., & Petersen, T. N. (2010). CPHmodels-3.0-remote homology modeling using structure-guided sequence profiles. Nucleic Acids Research, 38(SUPPL. 2), 576–581.
Krieger, E., Joo, K., Lee, J., Lee, J., Raman, S., Thompson, J., & Karplus, K. (2009). Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: four approaches that performed well in CASP8. Proteins: Structure, Function, and Bioinformatics, 77(SUPPL. 9), 114–122.
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. a., Immormino, R. M., Kapral, G. J., & Richardson, D. C. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D: Biological Crystallography, 66(1), 12–21.
Emsley, P., Lohkamp, B., Scott, W. G., & Cowtan, K. (2010). Features and development of Coot. Acta Crystallographica Section D: Biological Crystallography, 66(4), 486–501.
DeBeer, T. a. P., Berka, K., Thornton, J. M., & Laskowski, R. A. (2014). PDBsum additions. Nucleic Acids Research, 42(D1), 292–296. doi:10.1093/nar/gkt940.
McNicholas, S., Potterton, E., Wilson, K. S., & Noble, M. E. M. (2011). Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallographica Section D: Biological Crystallography, 67(4), 386–394.
Sun, F., Zhang, X. Z., Myung, S., & Zhang, Y. H. P. (2012). Thermophilic Thermotoga maritima ribose-5-phosphate isomerase RpiB: optimized heat treatment purification and basic characterization. Protein Expression and Purification, 82(2), 302–307.
Patchett, M. L., Neal, T. L., Schofield, L. R., Strange, R. C., Daniel, R. M., & Morgan, H. W. (1989). Heat treatment purification of thermostable cellulase and hemicellulase enzymes expressed in E. coli. Enzyme and Microbial Technology, 11(2), 113–115.
Gumerov, V. M., Rakitin, A. L., Mardanov, A. V., & Ravin, N. V. (2015). A novel highly thermostable multifunctional beta-glycosidase from Crenarchaeon acidilobus saccharovorans. Archaea, 2015, 978632.
Nguyen, Q. D., Rezessy-Szabó, J. M., Bhat, M. K., & Hoschke, Á. (2005). Purification and some properties of β-fructofuranosidase from Aspergillus niger IMI303386. Process Biochemistry, 40(7), 2461–2466.
Hayashi, S., Matsuzaki, K., Takasaki, Y., Ueno, H., & Imada, K. (1992). Purification and properties of b-fructofuranosidase from Aspergillus japonicus. Microbial Biotechnology, 8, 276–279.
Yanai, K., Nakane, A., Kawate, A., & Hirayama, M. (2001). Molecular cloning and characterization of the fructooligosaccharide-producing beta-fructofuranosidase gene from Aspergillus niger ATCC 20611. Bioscience Biotechnology and Biochemistry, 65, 766–773.
Bruins, M. E., Strubel, M., Van Lieshout, J. F. T., Janssen, A. E. M., & Boom, R. M. (2003). Oligosaccharide synthesis by the hyperthermostable beta-glucosidase from Pyrococcus furiosus: kinetics and modelling. Enzyme and Microbial Technology, 33(1), 3–11.
Zambelli, P., Fernandez-Arrojo, L., Romano, D., Santos-Moriano, P., Gimeno-Perez, M., Poveda, A., & Plou, F. J. (2014). Production of fructooligosaccharides by mycelium-bound transfructosylation activity present in Cladosporium cladosporioides and Penicilium sizovae. Process Biochemistry, 49(12), 2174–2180.
Holm, L., & Rosenström, P. (2010). Dali server: conservation mapping in 3D. Nucleic Acids Research, 38(SUPPL. 2), 545–549.
Hothorn, M., Van den Ende, W., Lammens, W., Rybin, V., & Scheffzek, K. (2010). Structural insights into the pH-controlled targeting of plant cell-wall invertase by a specific inhibitor protein. Proceedings of the National Academy of Sciences of the United States of America, 107(40), 17427–17432.
Bujacz, A., Jedrzejczak-Krzepkowska, M., Bielecki, S., Redzynia, I., & Bujacz, G. (2011). Crystal structures of the apo form of β-fructofuranosidase from Bifidobacterium longum and its complex with fructose. FEBS Journal, 278(10), 1728–1744.
Pouyez, J., Mayard, A., Vandamme, A. M., Roussel, G., Perpète, E. a., Wouters, J., & Michaux, C. (2012). First crystal structure of an endo-inulinase, INU2, from Aspergillus ficuum: discovery of an extra-pocket in the catalytic domain responsible for its endo-activity. Biochimie, 94(11), 2423–2430.
Deshpande, N., Wilkins, M. R., Packer, N., & Nevalainen, H. (2008). Protein glycosylation pathways in filamentous fungi. Glycobiology, 18(8), 626–637.
Shental-Bechor, D., & Levy, Y. (2008). Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proceedings of the National Academy of Sciences of the United States of America, 105(24), 8256–8261.
Acknowledgments
This work was supported by the CONACyT, México (No. 169048) and PROMEP. José Juan Virgen-Ortíz would also like to express his gratitude to CONACyT Mexico for his PhD fellowship No. 307333.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Virgen-Ortíz, J.J., Ibarra-Junquera, V., Escalante-Minakata, P. et al. Identification and Functional Characterization of a Fructooligosaccharides-Forming Enzyme from Aspergillus aculeatus . Appl Biochem Biotechnol 179, 497–513 (2016). https://doi.org/10.1007/s12010-016-2009-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-016-2009-8