
Abstract
Emergence of novel genome engineering technologies such as clustered regularly interspaced short palindromic repeat (CRISPR) has refocused attention on unresolved ethical complications of synthetic biology. Biosecurity concerns, deontological issues and human right aspects of genome editing have been the subject of in-depth debate; however, a lack of transparent regulatory guidelines, outdated governance codes, inefficient time-consuming clinical trial pathways and frequent misunderstanding of the scientific potential of cutting-edge technologies have created substantial obstacles to translational research in this area. While a precautionary principle should be applied at all stages of genome engineering research, the stigma of germline editing, synthesis of new life forms and unrealistic presentation of current technologies should not arrest the transition of new therapeutic, diagnostic or preventive tools from research to clinic. We provide a brief review on the present regulation of CRISPR and discuss the translational aspect of genome engineering research and patient autonomy with respect to the “right to try” potential novel non-germline gene therapies.
Similar content being viewed by others

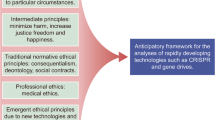
Introduction
Genetic engineering has been a laboratory tool for last few decades, but efficient technologies for precise targeting of the genome were not available. Recent advancement in bioengineering has given rise to a revolutionary genome editing technology. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)-associated system (Cas), was originally a bacterial defense system that came to researcher’s attention after its successful implementation in genome engineering and rapidly eclipsed conventional genome editing techniques. The first report of CRISPR cluster repeats was released in 1987 (Shino et al. 1987), but it wasn’t until 2013 that the power of CRISPR/Cas9 system for genome engineering in eukaryotic cells was demonstrated (Cong et al. 2013; Mali et al. 2013). According to the pioneers of CRISPR technology, the modern definition of genome engineering refers to targeted modification of the genome, its context (such as epigenetic markers) and its outputs (such as transcripts) (Hsu et al. 2014). This is a helpful reminder that genome editing technology such as CRISPR/Cas9 differs from classic genome editing methods in that manipulation of the genome was previously restricted to the use of Homologous Recombination (HR)-based techniques that rely on homology of the donor template and target region. Despite its significant efficacy in animal models, HR-based techniques had a low efficiency rate in human cells (Meissner et al. 2014).
One hallmark of the CRISPR/Cas9 system that was previously missing in other gene therapy techniques is the strong target recognition ability of the system, which is dictated by the Watson–Crick base pairing interactions of an RNA guide with its DNA target (Hsu et al. 2014). Target-specificity of CRISPR/Cas9 and the relative ease of use of this system has opened a variety of experimental opportunities for research, medicine and biotechnology. Since its primary application as a genome editing tool in 2013, it has been widely used in various cell lines and organisms including mice, rats, fruit flies, nematodes, arabidopsis, salamanders, frogs, and monkeys; crop plants e.g. rice, wheat, sorghum, and tobacco; and in different fungi, organoids, human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) (Doudna and Charpentier 2014).
Some major applications of the CRISPR/Cas9 system in research include creating germline mutations and generating transgenic animal models with significant efficiency and speed, transplantation-based in vivo models where stem cells or progenitor cells are modified ex vivo by CRISPR and transplanted into syngenic recipients, and the most popular adaptation where a direct in vivo delivery technique is used, such as microinjection of CRISPR via adeno-associated virus into tissue (Dow 2015). A proof-of-concept study in 2014 in a mouse model of cardiovascular disease suggested that gene therapy by CRISPR/Cas9 was able to permanently modify the mutant gene and restore the natural function of the gene product (Ding et al. 2014). Another recent study in non-human primates demonstrated that injection of a CRISPR/Cas9 system into a one-cell-stage embryo is able to simultaneously target two genes in one step, with high specificity and no recorded off-target mutagenesis (Niu et al. 2014).
According to a recent report in Nature, scientific publications on CRISPR outnumber those on any other gene editing technology, reaching over 700 in early 2014. Funding allocations and patent applications for CRISPR also indicate a significant shift towards this technology. Following early invention of CRISPR, at least four start-up/companies were founded with a major focus on therapeutic application of CRISPR, including Caribou Bioscience, Editas Medicine, CRISPR Therapeutics and Intellia Therapeutics (Ledford 2015).
CRISPR gene editing technology offers unrivalled opportunities in fighting genetic diseases and modifying genomes in human and other living organisms. The efforts of scientists in genetic engineering reached a peak when CRISPR appeared as a fast, straight-forward and low-cost technology accessible almost in any basic laboratory setting. However, from a consequentialist point of view the unknown risks and potential benefits pertaining to this powerful gene editing technology need a substantial investigation and an open discussion to enable a thorough assessment of scientific, ethical and societal aspects of this issue. Here, we review major commentaries on the classification of CRISPR/Cas9 genome editing technology in relation to the governance of synthetic biology more widely, its potential risks and benefits for public health and to provide a perspective on the transition of CRISPR from research to clinic.
Genetic engineering is the deliberate modification of an organism’s genetic material in order to alter or enhance its characteristics. New advances in technology are shifting towards not only editing genetic materials but also programming and creating a whole new set of genetic codes that equips the engineered organism with new capabilities that are not naturally occurring. Technologies such as CRISPR are narrowing the gap between editing and writing the genome. Whether it is a matter of marketing for industry or a matter of governance, in recent years genetic engineering has been regarded as a synthetic biology (synbio) tool. Table 1 describes three major buzzwords of synthetic biology often discussed in bioethics and social studies. Genome engineering techniques are classified under the category of synthetic biology; genome engineering can be targeted at prokaryotic or eukaryotic systems however gene therapy is a more precisely specified term that conveys a medical objective. Genetically modified organisms belong to the category of synbio, but does not involve research on or use in humans and is restricted to non-human living organisms.
CRISPR as a Synbio Tool
The European Commission’s synbio summit part one (Opinion on Synthetic Biology I Definition-2014), introduced synbio as “the application of science, technology and engineering to facilitate and accelerate the design, manufacture and/or modification of genetic materials in living organisms”. The summit categorised major synbio tools as design, construction and diagnostic tools whereas synbio methodology serves research on synthetic genomics and DNA synthesis, metabolic engineering, orthogonal biosystems/xenobiology and protocells. In June 2015, the second part of the summit on risk assessment methodologies and safety aspects of synbio classified genome editing technologies such as Multiplex Automated Genome Engineering (MAGE), Transcription activator-like effector nucleases (TALENs) and the (CRISPR)/Cas9 system as synbio tools (European commission 2015).
As an emerging field in itself, synbio is subject to much heated debate, and despite its high potential application in drug-discovery, development of medical therapeutics, diagnostic tools and improvement of bioproducts, its governance and regulatory strategies remain to be fully defined. Currently, the EU legal regulations which govern synbio, including biosafety and risk management, are previously existing biotechnology regulations on chemical/biological products and genetic modification research which are considered relevant to synbio. The debate and controversy around the frontiers of synthetic biology is partly due to the rapidly emerging techniques and methodologies that diverge from previously defined applications. Synbio applies engineering approaches to the study and design of living organisms. The major contribution of synbio to biomedicine is engineering biomolecules, genetic circuits and reprograming cells to modify pathologic pathways or enhance/reform their biological function in ways that previously did not exist in nature. Emerging applications of synbio include vaccine development, cancer treatment, prevention and treatment of infection, microbiome engineering, cell therapy and regenerative medicine (Ruder et al. 2011), biofuels and genome engineering.
One of the most exciting applications of synbio is the development of new diagnostic tools which offer easy-to-use, low-cost, out-of-laboratory solutions. One example is paper-based synthetic gene networks, where biosensors use a colour-changing protein to signal the presence of a pathogen e.g. strain-specific Ebola virus or antibiotic-resistant bacteria in the sample; the change of colour is visible to the naked eye and therefore applicable outside the laboratory facility (Pardee et al. 2014). Synthetic gene circuits such as clocks, oscillators, timers, counters and pattern-detectors are amongst the other inventions of synthetic biology that offer novel therapeutic approaches; the most complex circuits now take advantage of traceless inducers such as light and radio waves to regulate the gene expression (Ye and Fussenegger 2014). A recent proof-of-concept study, demonstrated that the brainwaves of a human subject could remotely switch on gene expression in mice. The brainwave turns on an LED implanted under the skin of the mice, and emission of light from the LED effectively switches on expression of light-sensitive genes which are engineered to respond to the light (Folcher et al. 2014). This mind-control concept uses electrical activity recorded by EEG (electroencephalogram) to control a device. The communication medium is the Brain-Computer-Interface (BCI) which is programmed to recognize a specific pattern; the BCI interprets different brain activity patterns that are associated to specific states of mind such as relaxation, and translates certain brain electrical patterns into machine actions. The brain activities integrated in BCI are not concrete “emotions”, but the same type of mental commands one executes to move an arm. In the machine learning process, it is both the machine and the patient that are self-trained to communicate. Although this experiment is only a proof-of-concept study, a mind-genetic interface could be a turning-point in therapeutic approaches, especially personalized medicine, as patients could modulate expression of certain proteins or synthetic products in a desired tissue and at needed time.
The number of inventions in the synbio domain is growing rapidly, as indicated by increasing number of research groups and publications (a synbio map that locates the laboratories and facilities worldwide concentrating on synbio was created in 2009 by the Woodrow Wilson International Center for Scholars http://www.synbioproject.org/sbmap/). Meanwhile, bioethicists and public health advocates have opened a dialogue on the harms, benefits, societal impacts and ethical hurdles of synbio research and development. Risk assessment in synbio is frequently discussed, but global guidelines that address this concern and promote harmonization are lacking, meaning that the debate continues to divide the community. One of the obstacles to synbio risk assessment stems from uncertainties of scientific advancement, as the risks and benefits of a potential treatment/device/product remains uncertain at the early developmental stage and real dimensions of the issue only emerge once the innovation reaches first-in-human (FIH) trials.
CRISPR and Human Gene Editing
Recent attempts by Chinese scientists to edit human embryos using CRISPR (Liang et al. 2015) caused much ethical and legal controversy. Committees of scientists and bioethicists have expressed their concern regarding the immature status of CRISPR in respect of its adverse effects, highlighting a need for thorough investigation of safety and efficacy issues prior to any attempt for engineering the human genome (Baltimore et al. 2015). Likewise, the US National Institutes of Health (NIH) reaffirmed that NIH will not fund any use of gene-editing technologies in human embryos in accordance with the Dickey-Wicker amendment (1996), which prohibits the use of federal funds for creating, destroying, or knowingly injuring human embryos (Collins 2015). The US Food and Drug Administration (FDA) works as the final arbiter of the clinical application of gene therapy, but such decisions are subject to review by the Recombinant DNA Advisory Committee (RAC) of NIH. In 2013, in its revisions on guidelines for research involving recombinant/synthetic nucleic acid molecules, NIH stated that “RAC will not at present entertain proposals for germ line alterations but will consider proposals involving somatic cell gene transfer” (Department of health and human services NIH 2013). Interference with germline genetic makeup has been a socially sensitive issue since the dawn of genetic engineering as it escalates a biological intervention to a highly ethical perspective. For instance article 1 of the Universal Declaration on the Human Genome and Human Rights (1997) declares that “the human genome underlies the fundamental unity of all members of the human family, as well as the recognition of their inherent dignity and diversity. In a symbolic sense, it is the heritage of humanity” (UNESCO 1997). The declaration of UNESCO classifies the human genome as a world heritage, which is inherently subject to protection and conservation for future generations. Although UNESCO considers the integrity of the human genome that evolves, carries mutations and expresses different potentialities in each individual, It remains vague whether genetic deficiencies and impairments that cause serious diseases are considered as variations of human genome with an evolutionary purpose and therefore subject to protection and conservation, or whether they are biological errors that could ethically be corrected by means of technology. Their statement begs the question: what is protection or conservation of the human genome? If we are able to correct biological errors, is it not our moral responsibility to future generations to do so?
The European Commission view on gene therapy is reflected in Directive 2009/120/EC which refers to gene therapy, somatic cell therapy and tissue engineering as advanced therapy. However, due to technical complications of medical devices and the interdisciplinary aspect of advanced therapy, a special committee was formed to organize a case-by-case investigation for emerging technologies that might fall under advanced therapies. The Committee for Advanced Therapies (CAT) is the centralised marketing authorisation body that is responsible for evaluation of new products of advanced therapy and technical regulation of respective technologies. According to CAT, a gene therapy medicinal product “(a) contains an active substance which contains or consists of a recombinant nucleic acid used in or administered to human beings with a view to regulating, repairing, replacing, adding or deleting a genetic sequence; (b) its therapeutic, prophylactic or diagnostic effect relates directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence” (European Council 2009a). CRISPR/Cas9’s use of adeno-associated viral vectors (AAV) for in vivo delivery appears to fit well with this definition. Although gene therapy in somatic cells seems to have the conditional approval of European Union legislation, any interference in genetic material of the germline is prohibited as indicated in the Convention on Human Rights and Biomedicine of the European Council: The treaty (No. 164) allows genetic engineering only for preventive, diagnostic or therapeutic reasons and only where it does not aim to change the genetic make-up of a person’s descendants (European Council 1997). Directive 2001/20/EC (on the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use), also forbids gene therapy clinical trials that may result in modification of a germline genetic identity (European Council 2009b), however, the directive overlooks the possibility of using gene therapy on the germline to prevent the inheritance of a genetic disease. Finally, Directive 98/44/EC (on the legal protection of biotechnological inventions) exempts processes for modifying the germline genetic identity of human beings, human cloning and the use of human embryos for commercial and industrial purposes from patentability on the basis that “inventions shall be considered unpatentable where their commercial exploitation would be contrary to public order or morality” (European Council 1998). Of course, it could and should be argued that using germline genetic modification to prevent the transmission of serious genetic disease to future generations is clearly not contrary to public order or morality.
CRISPR raises issues across a dynamic spectrum of science, ethics and policy. While we acknowledge the principle of “do no harm”, A consistent dialogue between science and ethics can balance the position of such technologies in political decision and law making. However as a perquisite of democratic legislation we need to effectively involve the public voice in this procedure. One risk concerning the public perception of cutting-edge technologies, and in particular those related to human health, is the validity of the information accessible to the public. Mass media sometimes misrepresents science to the public, and the fine line between science and fiction should be taken seriously when discussing genome editing to avoid hype surrounding its medical implications, and misplaced fear regarding potential risks. On the other hand, one may argue that scientists are not elected by people and may not necessarily represent the values of the society (Sarewitz 2015); whether their technical knowledge qualifies them for decision making remains subjective. A transparent policy where realistic advantage and disadvantage of such technologies are communicated with the public will certainly serve the purpose. In Table 2 we summarize some of frequently addressed benefits and risks of synbio tools and in particular genome-engineering techniques in a socio-ethical context.
CRISPR from Lab to Clinic
From a technical point of view, CRISPR and other gene editing tools have a long way to go before they could be used to alter the human germline for preventive purposes; even if safety needs are met, what restricts germline gene therapy is our limited knowledge of genetics. In other words, our current understanding of genetic disorders restricts CRISPR and such technologies to a small number of diseases where both parents carry a mutant allele for a dominant disease such as Huntington’s disease, or when both parents have the same autosomal recessive disease e.g. cystic fibrosis. Even in such rare cases, other well-established preventive strategies such as preimplantation genetic diagnosis (PGD) or fetal genetic testing would currently be medically preferable to the uncertainties of germline editing (Greely 2015). On the contrary, somatic cell therapy appears to be a less controversial starting point for CRISPR since it does not raise the concerns of heritability of genetic modifications; however it is still subject to massive safety issues. In Table 3 we summarise the main controversies on germline vs somatic cell therapy. Somatic cell therapy does not induce heritable genetic modification, so where applicable, patients who have failed all other treatments should be granted the right to try novel technologies such as CRISPR, even if the chances of therapeutic efficacy are almost non-existent. Although CRISPR reignites the old controversy of germline modification, the real challenge we face is not legislation of germline modification, but to promote timely exploitation of such technologies to move forward with long awaited gene therapy. In a ground-breaking report released during preparation of this review, TALEN- a genetic research technique-successfully cured leukaemia in a 1-year-old girl; the treatment was not a clinical trial and had only been tested in animal models, yet researchers were able to obtain a special permission to try it on the baby in whom all other treatments had failed (Reardon 2015). Such innovative cases demonstrate the life-saving potential of novel genome editing technology, and may pave the way for a revised assessment of clinical trial paths for gene therapy.
In 2015, the debate on genome editing technology and in particular CRISPR reached several milestones: in January 2015 NAPA meeting on new prospects in genome engineering led to a call for a moratorium on human germline genome modification in March. The first report of a CRISPR application for genetic modification in non-viable human embryos was released in April 2015. Simultaneously, the NIH reaffirmed that it will not fund any use of gene-editing technologies in human embryos (Collins 2015). In September 2015, the German National Academy of Sciences (Leopoldina) endorsed a call for an international moratorium on all kinds of genome editing interventions in human germline that affect offsprings’ genomes. In October 2015, the International Bioethics Committee (IBC) of UNESCO declared genetic modification should be admitted only for prevention, diagnostic and therapeutic purposes without transmission of any artificial genetic alteration to the next generation (International bioethics committee of UNESCO 2015). However, the position of IBC needs further interpretation since some (if only a few) genetic diseases such as Huntington’s disease can be prevented in the next generation by replacing the faulty genes in the germline of their parents. If genetic alterations could be transmitted to future generations purely for therapeutic reasons, this would prevent future children and adults from developing serious diseases without requiring each of them to undergo risky and costly gene therapy individually. Does this preventive rationale exempt such cases from the IBC ban on germline interventions? Finally in December 2015 the International Summit on Human Gene Editing-Washington, DC was co-hosted by the US National Academy of Sciences, the National Academy of Medicine, the Chinese Academy of Sciences, and the Royal Society of the UK. The summit released an expert statement of the use of genome editing technology as a basic research tool and also genome editing at the somatic and germline level. The summit also called for the hosts to take the lead and establish an international forum to maintain an ongoing discussion on the clinical application of genome editing technology, and harmonize the regulations and formulations of guidelines among nations.
To enable effective translation of CRISPR-like technologies to the clinic, several major challenges of safety and efficiency must be overcome, as for any other advanced technology. The lack of a global benchmark for human genome editing, particularly in regard to new technologies, further complicates matters at the translational level. There are currently no clinical trials of CRISPR as a gene therapy technique, but in a recent clinical trial using ZFN gene editing technique on patients with HIV, investigators were able to introduce a disease-resistance allele into patient’s immune cells to keep the viral load at bay. CRISPR is anticipated to have higher targeting precision than ZFN and therefore holds promises for such applications.
Although researchers are pushing scientific boundaries by reprogramming cells and organisms, synbio translational research progresses slowly. Slow transition of novel technologies such as CRISPR to the clinic is partly due to the shortage of translational expertise, the long clinical trial pathway, inefficient regulatory affairs and unresolved issues in the ethics of FIH trials. Accelerating translational research requires pragmatic solutions which simultaneously protect patients’ rights and encourage scientists to move forward with their discoveries. The FDA’s 2006 guideline on investigational new drugs (IND) introduced phase 0 clinical trials to speed up development of new treatments. Phase 0 trials are performed early in phase 1 on a small number of participants for a limited exposure time and a limited dose of the drug; a phase 0 trial has no therapeutic intentions and is a primary go/no-go decision making process based on human data instead of traditional animal data (LoRusso 2009; U.S Food and Drug Administration 2006).The European Medicines Agency (EMA) has also reformed its early phase I (microdose) clinical trial regulation to promote an early distinction of promising products from others (European Medicines Agency 2006). What makes phase 0 trials ethical is their hypothetical value in shortening the clinical trial path and the potential benefit of this for the patient population who would be recruited in later phases of the study.
While phase 0 and phase 1 trials serve no therapeutic purpose, one may reiterate a “Right to try” for patient groups whose life may depend on new therapeutics which are not yet approved. Questions can be raised about whether such patients can freely consent to participate in such trials given their dire situation and the risk of the therapeutic misconception; nonetheless, any new CRISPR therapy will have to be tested on humans at some stage, and denying patients at least the chance to contribute towards testing a new therapy for other patients with their condition would not be logical or ethical. Although it is a new technology, the same ethical principles apply as in the case of early-phase cancer trials.
Once CRISPR therapies reach the clinical trial stage, patients with life-limiting genetic disorders may become the participant group of choice for research, just as terminally ill cancer patients are currently favoured in clinical trials of highly toxic new therapies. Research ethics committees (RECs) have to rely on experimental risk/benefit assessments to proceed with FIH trials, leaving patients with no input in such a decision making process; however, putting patients in ethics committees and actively involving their perspective in such assessments particularly those related to novel technologies provides an invaluable source of information on risks and benefits as seen by well-informed patients (Shaw and Elger 2014). The practice of involving patients in the ethics committee and decision making process undoubtedly requires a consistent and reliable information exchange between patient and the physician. As the informative model of physician-patient relationship suggests the physician informs the patient of their state of the disease, present diagnosis and available interventions and also provides the information of the nature and probability of the risks and benefits of the treatments, the physician is equally responsible to present the uncertainties of the interventions. The patient has the freedom to exercise control over this decision making process and choose the intervention that is the best fit to his or her conditions (Emanuel and Emanuel 1992).
Despite its sophisticated methodology, genome engineering is a concept easily grasped by the public, as evidenced by the staggering number of news articles, social media discussions, books and movies on its advantages and risks, although the media may succeed to create hype in genome engineering, yet not involving the public voice in decision making is an underestimation of the public understanding.
Concluding Remarks
A precautionary approach towards CRISPR translational research is rational, but as is the case with some other technologies; fear of its failure or misuse may restrict it to basic science research as an exploratory lab technique. Participation in genome editing research where it does not interfere with the germline and is hence not transmittable to future generations should remain a personal choice for patients whose life expectancy may not survive the long clinical trial path. Clinical use of genome editing technology on somatic cells does not raise ethical issues related to the inheritance of the genetic alterations; therefore, when regulatory needs and safety concerns are met, somatic gene editing by the use of CRISPR should be incorporated into trial designs and clinical applications. As the expert statement of the international summit of December 2015 declared, germline editing technology could be of significant benefit in pre-clinical and clinical uses. However, several factors necessitate the current restriction on germline editing technology such as CRISPR. These include the current state of uncertainty on safety and security issues, the paradox of individual and future generation needs, and the possibility of permanent alteration of the human genome in ways that increase social inequality, among other ethical considerations. However, this declaration is not, and should not be permanent, as CRISPR holds great promises.
References
Baltimore, D., Berg, P., Botchan, M., Carroll, D., Charo, R. A., Church, G., et al. (2015). Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science, 348(6230), 36–38. doi:10.1126/science.aab1028.
Collins, F. S. (2015). The NIH director. Statement on NIH funding of research using gene-editing technologies in human embryos. http://www.nih.gov/about-nih/who-we-are/nih-director/statements/statement-nih-funding-research-using-gene-editing-technologies-human-embryos. Accessed 07 Dec 2015.
Cong, L., Ran, F. A., Cox, D., Lin, S., Barretto, R., Habib, N., et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823.
Dana, G. V., Kuiken, T., Rejeski, D., & Snow, A. A. (2012). Synthetic biology: Four steps to avoid a synthetic-biology disaster. Nature. doi:10.1038/483029a.
Department of health and human services NIH. (2013). Revisions of the NIH guidelines for research involving recombinant or synthetic nucleic acid molecules. http://osp.od.nih.gov/sites/default/files/NIH_Guidelines_0.pdf. Accessed 07 Dec 2015.
Ding, Q., Strong, A., Patel, K. M., Ng, S. L., Gosis, B. S., Regan, S. N., et al. (2014). Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circulation Research, 115(5), 488–492. doi:10.1161/CIRCRESAHA.115.304351.
Doudna, J. A., & Charpentier, E. (2014). Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science, 346(6213), 1258096. doi:10.1126/science.1258096.
Dow, L. E. (2015). Modeling disease in vivo with CRISPR/Cas9. Trends in Molecular Medicine, 21(10), 609–621. doi:10.1016/j.molmed.2015.07.006.
Emanuel, E. J., & Emanuel, L. L. (1992). Four models of the physician-patient relationship. JAMA, 267(16), 2221–2226.
European Commission. (2014). Opinion on synthetic biology I definition. http://ec.europa.eu/health/scientific_committees/emerging/docs/scenihr_o_044.pdf. Accessed 07 Dec 2015.
European Commission. (2015). Synthetic biology II—Risk assessment methodologies and safety aspects, opinion. http://ec.europa.eu/health/scientific_committees/emerging/docs/scenihr_o_048.pdf. Accessed 07 Dec 2015.
European Council. (1997). Convention for the protection of human rights and dignity of the human being with regard to the application of biology and medicine: Convention on human rights and biomedicine. https://rm.coe.int/CoERMPublicCommonSearchServices/DisplayDCTMContent?documentId=090000168007cf98. Accessed 07 Dec 2015.
European Council. (1998). Directive 98/44/EC of the European Parliament and of the Council of 6 July 1998 on the legal protection of biotechnological inventions http://eur-lex.europa.eu/legal-content/EN/NOT/?uri=CELEX:31998L0044. Accessed 07 Dec 2015.
European Council. (2009a). Directive 2001/20/ec of the european parliament and of the council of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the member states relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Official Journal of the European Commuinities, OJ L, 121, 34–44.
European Council. (2009b). Commission directive 2009/120/EC of 14 September 2009 amending directive 2001/83/EC of the European Parliament and of the council on the community code relating to medicinal products for human use as regards advanced therapy medicinal products. Official Journal of the European Commuinities, L242, 3–12.
European Council. (2009c). Contained use of genetically modified micro-organisms (GMMs) http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=URISERV:sa0015. Accessed 07 Dec 2015.
European Medicines Agency. (2006). Concept paper on the development of a CHMP guideline on the non-clinical requirements to support early phase I clinical trials with pharmaceutical compounds. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/10/WC500003979.pdf. Accessed 07 Dec 2015.
Folcher, M., Oesterle, S., Zwicky, K., Thekkottil, T., Heymoz, J., Hohmann, M., et al. (2014). Mind-controlled transgene expression by a wireless-powered optogenetic designer cell implant. Nature Communications, 5, 5392. doi:10.1038/ncomms6392.
Food and drug administration center for biologics evaluation and research. (1998). Guidance for industry: Guidance for human somatic cell therapy and gene therapy. http://www.fda.gov/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm072987.htm#i. Accessed 07 Dec 2015.
Greely, H. (2015). Law and biosciences blog of science, CRISPR-Cas9, and Asilomar. https://law.stanford.edu/2015/04/04/of-science-crispr-cas9-and-asilomar/. Accessed 07 Dec 2015.
Hsu, P. D., Lander, E. S., & Zhang, F. (2014). Development and applications of CRISPR-Cas9 for genome engineering. Cell, 157(6), 1262–1278. doi:10.1016/j.cell.2014.05.010.
International bioethics committee of UNESCO (2015). (2015). Report of the IBC on updating its reflection on the human genome and human rights. http://unesdoc.unesco.org/images/0023/002332/233258E.pdf. Accessed 07 Dec 2015.
Ledford, H. (2015). CRISPR, the disruptor. Nature, 522(7554), 20–24. doi:10.1038/522020a.
Liang, P., Xu, Y., Zhang, X., Ding, C., Huang, R., Zhang, Z., et al. (2015). CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein and Cell, 6(5), 363–372. doi:10.1007/s13238-015-0153-5.
LoRusso, P. M. (2009). Phase 0 clinical trials: an answer to drug development stagnation? Journal of Clinical Oncology, 27(16), 2586–2588. doi:10.1200/JCO.2008.21.5798.
Mali, P., Yang, L., Esvelt, K. M., Aach, J., Guell, M., DiCarlo, J. E., et al. (2013). RNA-guided human genome engineering via Cas9. Science, 339(6121), 823–826. doi:10.1126/science.1232033.
Meissner, T. B., Mandal, P. K., Ferreira, L. M., Rossi, D. J., & Cowan, C. A. (2014). Genome editing for human gene therapy. Methods in Enzymology, 546, 273–295. doi:10.1016/B978-0-12-801185-0.00013-1.
Niu, Y., Shen, B., Cui, Y., Chen, Y., Wang, J., Wang, L., et al. (2014). Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell, 156(4), 836–843. doi:10.1016/j.cell.2014.01.027.
Pardee, K., Green, A. A., Ferrante, T., Cameron, D. E., DaleyKeyser, A., Yin, P., et al. (2014). Paper-based synthetic gene networks. Cell, 159(4), 940–954. doi:10.1016/j.cell.2014.10.004.
Reardon, S. (2015). Leukaemia success heralds wave of gene-editing therapies. Accessed 06 Nov 2015.
Ruder, W. C., James, T. L., & Collins, J. J. (2011). Synthetic biology moving into the clinic. Science, 333(6047), 1248–1252. doi:10.1126/science.1206843
Sarewitz, D. (2015). CRISPR: Science can’t solve it. Nature Comments, 522(7557), 413–414.
Shaw, D., & Elger, B. (2014). Putting patients on research ethics committees. Journal of the Royal Society of Medicine, 107(8), 304–307.
Shino, Y., Shinagawa, H., Makino, K., Amemura, M., & Nakata, A. (1987). Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. Journal of Bacteriology, 169, 5429–5433.
UNESCO. (1997). Universal declaration on the human genome and human rights. http://portal.unesco.org/en/ev.php-URL_ID=13177&URL_DO=DO_TOPIC&URL_SECTION=201.html. Accessed 07 Dec 2015.
U.S Food and Drug Administration. (2006). Guidance for industry, investigators, and reviewers exploratory in studies. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm078933.pdf. Accessed 07 Dec 2015.
Ye, H., & Fussenegger, M. (2014). Synthetic therapeutic gene circuits in mammalian cells. FEBS Letters, 588(15), 2537–2544. doi:10.1016/j.febslet.2014.05.003.
Acknowledgments
This research project was funded by the University of Basel and the ‘Swiss National Science Foundation’ under project Grant No. PDFMP3_137194/1.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Heidari, R., Shaw, D.M. & Elger, B.S. CRISPR and the Rebirth of Synthetic Biology. Sci Eng Ethics 23, 351–363 (2017). https://doi.org/10.1007/s11948-016-9768-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11948-016-9768-z