
Abstract
Hereditary central nervous tumor syndromes are a varied group of conditions that include neurofibromatosis type 1 and 2, tuberous sclerosis, Von Hippel-Lindau disease, and Cowden, Turcot, and Gorlin syndromes. The responsible genes have been identified in most of these disorders. These genes typically act as tumor suppressor genes, maintain normal cellular function and homeostasis, and regulate cell growth and differentiation. Familial central nervous system tumors are mostly inherited as autosomal dominant traits and involve germline mutations. Neoplastic development occurs when a somatic mutation inactivates the second allele. These patients also present unique challenges for their management. This review highlights the clinical manifestations, molecular genetics, pathophysiology, and current treatment options of these disorders with a focus on neuro-oncologic manifestations of the diseases.
Similar content being viewed by others

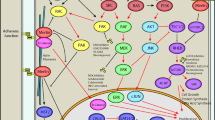

References and Recommended Reading
Gutmann DH: The neurofibromatoses: when less is more. Hum Mol Genet 2001, 10:747–755.
Friedman JM: Epidemiology of neurofibromatosis type 1. Am J Med Genet 1999, 89:1–6.
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988, 45:575–578.
Quigg M, Rust RS, Miller JQ: Clinical findings of the phakomatoses: neurofibromatosis. Neurology 2006, 66:E23–24.
Hofman KJ, Harris EL, Bryan RN, Denckla MB: Neurofibromatosis type 1: the cognitive phenotype. J Pediatr 1994, 124:S1–8.
Rosser TL, Vezina G, Packer RJ: Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology 2005, 64:553–555.
Rawal A, Yin Q, Roebuck M, et al.: Atypical and malignant peripheral nerve-sheath tumors of the brachial plexus: report of three cases and review of the literature. Microsurgery 2006, 26:80–86.
Bausch B, Borozdin W, Neumann HP: Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N Engl J Med 2006, 354:2729–2731.
Jahraus CD, Tarbell NJ: Optic pathway gliomas. Pediatr Blood Cancer 2006, 46:586–596.
Ogura, T, Adachi J, Nishikawa R, et al.: Synchronous optic and pineal pilocytic astrocytomas in a paediatric patient with neurofibromatosis type 1. Pediatr Neurosurg 2004, 40:301–305.
Deliganis AV, Geyer JR, Berger MS: Prognostic significance of type 1 neurofibromatosis (von Recklinghausen Disease) in childhood optic glioma. Neurosurgery 1996, 38:1114–1148; discussion 1118–1119.
King A, Listernick R, Charrow J, et al.: Optic pathway gliomas in neurofibromatosis type 1: the effect of presenting symptoms on outcome. Am J Med Genet 2003, 122:95–99.
Opocher E, Kremer LC, Da Dalt L, et al.: Prognostic factors for progression of childhood optic pathway glioma: a systematic review. Eur J Cancer 2006, 42:1807–1816.
Listernick R, Louis DN, Packer RJ, Gutmann DH: Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol 1997, 41:143–149.
Listernick R, Ferner RE, Piersall L, et al.: Late-onset optic pathway tumors in children with neurofibromatosis 1. Neurology 2004, 63:1944–1946.
Astrup J: Natural history and clinical management of optic pathway glioma. Br J Neurosurg 2003, 17:327–335.
Walker D: Recent advances in optic nerve glioma with a focus on the young patient. Curr Opin Neurol 2003, 16:657–664.
Widemann BC, Salzer WL, Arceci RJ, et al.: Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol 2006, 24:507–516.
Perilongo G, Moras P, Carollo C, et al.: Spontaneous partial regression of low-grade glioma in children with neurofibromatosis-1: a real possibility. J Child Neurol 1999, 14:352–356.
Broniscer A, Gajjar A, Bhargava R, et al.: Brain stem involvement in children with neurofibromatosis type 1: role of magnetic resonance imaging and spectroscopy in the distinction from diffuse pontine glioma. Neurosurgery 1997, 40:331–337; discussion 337–338.
Packer RJ, Ater J, Allen J, et al.: Carboplatin and vincristine chemotherapy for children with newly diagnosed progressive low-grade gliomas. J Neurosurg 1997, 86:747–754.
Grill J, Couanet D, Cappelli C, et al.: Radiation-induced cerebral vasculopathy in children with neurofibromatosis and optic pathway glioma. Ann Neurol 1999, 45:393–396.
Hamaratoglu F, Willecke M, Kango-Singh M, et al.: The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol 2005, 27–36.
Hirokawa, Y, Tikoo A, Huynh J, et al.: A clue to the therapy of neurofibromatosis type 2: NF2/merlin is a PAK1 inhibitor. Cancer J 2004, 10:20–26.
Baser ME, Friedman JM, Aeschliman D, et al.: Predictors of the risk of mortality in neurofibromatosis 2. Am J Hum Genet 2002, 71:715–723.
Evans DG, Huson SM, Donnai D, et al.: A clinical study of type 2 neurofibromatosis. Q J Med 1992, 84:603–618.
Parry DM, Eldridge R, Kaiser-Kupfer MI, et al.: Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 1994, 52:450–461.
Evans DG, Baser ME, O’Reilly B, et al.: Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg 2005, 19:5–12.
Brennan JW, Rowed DW, Nedzelski JM, Chen JM: Cerebrospinal fluid leak after acoustic neuroma surgery: influence of tumor size and surgical approach on incidence and response to treatment. J Neurosurg 2001, 94:217–223.
Evans DG, Birch JM, Ramsden RT, et al.: Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet 2006, 43:289–294.
Ho SY, Kveton JF: Rapid growth of acoustic neuromas after stereotactic radiotherapy in type 2 neurofibromatosis. Ear Nose Throat J 2002, 81:831–833.
Lenarz T, Lim HH, Reuter G, et al.: The auditory midbrain implant: a new auditory prosthesis for neural deafnessconcept and device description. Otol Neurotol 2006, 27:838–843.
Mayfrank L, Mohadjer M, Wullich B.: Intracranial calcified deposits in neurofibromatosis type 2. A CT study of 11 cases. Neuroradiology 1990, 32:33–37.
Crino PB, Nathanson KL, Henske EP: The tuberous sclerosis complex. N Engl J Med 2006, 355:1345–1356.
Roach ES, Gomez MR, Northrup H: Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998, 13:624–628.
Thiele EA: Managing epilepsy in tuberous sclerosis complex. J Child Neurol 2004, 19:680–686.
Curatolo P, Bombardieri R, Verdecchia M, Seri S: Intractable seizures in tuberous sclerosis complex: from molecular pathogenesis to the rationale for treatment. J Child Neurol 2005, 20:318–325.
Goh S, Butler W, Thiele EA: Subependymal giant cell tumors in tuberous sclerosis complex. Neurology 2004, 63:1457–1461.
Sutton LN: Current management of low-grade astrocytomas of childhood. Pediatr Neurosci 1987, 13:98–107.
Franz DN, Leonard J, Tudor C, et al.: Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006, 59:490–498.
Bader RS, Chitayat D, Kelly E, et al.: Fetal rhabdomyoma: prenatal diagnosis, clinical outcome, and incidence of associated tuberous sclerosis complex. J Pediatr 2003, 143:620–624.
Seizinger BR, Rouleau GA, Ozelius LJ, et al.: Von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988, 332:268–269.
Shuin T, Yamasaki I, Tamura K, et al.: Von Hippel-Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol 2006, 36:337–343.
Linehan WM, Lerman MI, Zbar B: Identification of the von Hippel-Lindau (VHL) gene. Its role in renal cancer. JAMA 1995, 273:564–570.
Glasker S: Central nervous system manifestations in VHL: genetics, pathology and clinical phenotypic features. Fam Cancer 2005, 4:37–42.
Richard S, Campello C, Taillandier L, et al.: Haemangioblastoma of the central nervous system in von Hippel-Lindau disease. French VHL Study Group. J Intern Med 1998, 243:547–553.
Wanebo JE, Lonser RR, Glenn GM, Oldfield EH: The natural history of hemangioblastom as of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg 2003, 98:82–94.
Dwarakanath S, Suri A, Sharma BS, Mehta VS: Intracranial hemangioblastomas: an institutional experience. Neurol India 2006, 54:276–278.
Conway JE, Chou D, Clatterbuck RE, et al.: Hemangioblastomas of the central nervous system in von Hippel-Lindau syndrome and sporadic disease. Neurosurgery 2001, 48:55–62.
Weil RJ, Lonser RR, DeVroom HL, et al.: Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg 2003, 98:95–105.
Escudier B, Eisen T, Stadler WM, et al.: Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007, 356:125–134.
Motzer RJ, Bukowski RM: Targeted therapy for metastatic renal cell carcinoma. J Clin Oncol 2006, 24:5601–5608.
Singh AD, Shields CL, Shields JA: von Hippel-Lindau disease. Surv Ophthalmol 2001, 46:117–142.
Manski TJ, Heffner DK, Glenn GM, et al.: Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA 1997, 277:1461–1466.
Kleihues, P, Schauble B, Zur Hausen A, et al.: Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol 1997, 150:1–13.
Olivier M, Goldgar De, Sodha N, et al.: Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003, 63:6643–6650.
Frebourg T, Abel A, Bonaiti-Pellie C, et al.: Li-Fraumeni syndrome: update, new data and guidelines for clinical management. Bull Cancer 2001, 88:581–587.
Taylor MD, Mainprize TG, Rutka JT: Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery 2000, 47:888–901.
Gorlin RJ: Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987, 66:98–113.
Vortmeyer AO, Stavrou T, Selby D, et al.: Deletion analysis of the adenomatous polyposis coli and PTCH gene loci in patients with sporadic and nevoid basal cell carcinoma syndrome-associated medulloblastoma. Cancer 1999, 85:2662–2667.
Raffel C: Medulloblastoma: molecular genetics and animal models. Neoplasia 2004, 6:310–322.
Albright AL, Wisoff JH, Zeltzer PM, et al.: Effects of medulloblastoma resections on outcome in children: a report from the Children’ Cancer Group. Neurosurgery 1996, 68:265–271.
Hubbard JL, Scheithauer BW, Kispert DB, et al.: Adult cerebellar medulloblastomas: the pathological, radiographic, and clinical disease spectrum. J Neurosurg 1989, 70:536–544.
Packer RJ, Sutton LN, Elterman R, et al.: Outcome for children with medulloblastoma treated with radiation and cisplatin, CCNU, and vincristine chemotherapy. J Neurosurg 1994, 81:690–698.
Louis DN, von Deimling A: Hereditary tumor syndromes of the nervous system: overview and rare syndromes. Brain Pathol 1995, 5:145–151.
Hamilton SR, Liu B, Parsons RE, et al.: The molecular basis of Turcot’ syndrome. N Engl J Med 1995, 332:839–847.
Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP: PTEN is essential for embryonic development and tumour suppression. Nat Genet 1998, 19:348–355.
Backman S, Stambolic V, Mak T: PTEN function in mammalian cell size regulation. Curr Opin Neurobiol 2002, 12:516–522.
Chow LM, Baker SJ: PTEN function in normal and neoplastic growth. Cancer Lett 2006, 241:184–196.
Lyons CJ, Wilson CB, Horton JC: Association between meningioma and Cowden’ disease. Neurology 1993, 43:1436–1437.
Tuli S, Provias JP, Bernstein M: Lhermitte-Duclos disease: literature review and novel treatment strategy. Can J Neurol Sci 1997, 24:155–160.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hottinger, A.F., Khakoo, Y. Update on the management of familial central nervous system tumor syndromes. Curr Neurol Neurosci Rep 7, 200–207 (2007). https://doi.org/10.1007/s11910-007-0031-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11910-007-0031-5