
Abstract
The heart and the kidneys are the most commonly involved organs in systemic amyloidosis. Cardiac involvement is associated with an increased morbidity, treatment intolerance, and poorer overall survival. The most common types of amyloidosis that are associated with cardiac involvement include light chain (AL) amyloidosis and transthyretin (TTR) amyloidosis (both mutant and wild type). The traditional first-line treatment for AL amyloidosis includes alkylator-based chemotherapy or high-dose melphalan followed by autologous stem cell transplantation (ASCT). Novel agents, including proteasome inhibitors, immunomodulators, and monoclonal antibodies, have shown promising activity in both frontline and relapsed settings. Orthotopic heart transplantation (OHT) followed by ASCT has led to superior outcomes compared to OHT alone. Orthotopic liver transplantation (OLT) is the first-line treatment for TTR amyloidosis. However, progression of cardiac amyloidosis after OLT is often noted due to deposition of wild TTR. Combined OLT and OHT also has a role in treatment and leads to superior outcomes in carefully selected candidates. Pharmacologic agents, including diflunisal, tafamidis, small interfering ribonucleic acid, and doxycycline, have shown promising activity in stabilizing TTR from misfolding into fibrils and are being actively investigated. Best supportive care and management of heart failure symptoms with diuretics are a mainstay of treatment in all cardiac amyloidosis subtypes. Robust data on the benefit of angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, or beta blockers in amyloid cardiomyopathy is lacking.
Similar content being viewed by others


Introduction
Amyloidosis constitutes a group of protein folding disorders, characterized by the deposition of insoluble protein aggregates in tissues, leading to end-organ damage. The amyloid protein is deposited as rigid non-branching fibrils and is identified by apple-green birefringence under polarized microscopy upon staining with Congo red (CR) dye [1]. The heart and the kidneys are the most common organs involved in systemic amyloidosis. Cardiac involvement is determined by the presence of clinical and/or laboratory parameters including elevation of cardiac biomarkers (NT-proBNP, cardiac troponin) coupled with echocardiographic or magnetic resonance (MR) imaging evidence of amyloidosis and a positive non-cardiac biopsy for CR staining [2]. Rarely, endomyocardial biopsy (EMB) is needed to confirm cardiac involvement, as 85 % or more of patients have evidence for amyloid deposition in fat or bone marrow biopsy. Histologically, cardiac amyloidosis is characterized by extracellular amyloid deposition in the myocardium with or without vascular involvement [3]. The appearance on cardiac MR imaging is shown in Fig. 1. Amyloid infiltration of the heart results in biventricular wall thickening without dilation of the ventricles (Fig. 2). This in turn leads to increased atrial pressure, resulting in bi-atrial dilation despite thickening of the atrial walls [4]. Amyloid cardiomyopathy should be suspected in all patients presenting with heart failure and preserved ejection fraction.

Cardiac magnetic resonance imaging. A Early gadolinium phase showing asymmetric hypertrophy of the interventricular septum compared with the ventricle free wall. B Short-axis view showing transmural late gadolinium enhancement (arrow) typically seen in AL amyloidosis (LV left ventricle, RV right ventricle)

Gross specimen of the heart (cross-sectional view), affected by amyloid cardiomyopathy
The focus of this paper is the management and newer treatment strategies in cardiac amyloidosis with a brief review of the pathogenesis and clinical features of the different subtypes of cardiac amyloidosis.
Immunoglobulin Light Chain (AL) Amyloidosis
AL amyloidosis or primary systemic amyloidosis is the most common form of amyloidosis in the USA [5]. The estimated annual incidence is 3 per million person-years [6]. Pathologically, AL amyloidosis is characterized by a clonal population of plasma cells in the bone marrow, which produces amyloidogenic monoclonal light chains. Amyloidosis can arise de novo or in association with another plasma cell dyscrasia. The heart is affected in about 50 % of patients with AL amyloidosis [4]. The median age of presentation is 59 years. The median OS from diagnosis in Mayo stages I, II, III, and IV (defined in Table 1) are 94, 40, 14, and 6 months respectively [7]. The median OS from onset of CHF is around 9 months [8]. Periorbital ecchymosis and macroglossia are present in 12 and 27 % of patients, respectively [8].
Senile Systemic Amyloidosis (Wild-Type ATTR)
Senile systemic amyloidosis (SSA) is caused by the deposition of amyloid fibrils derived from wild-type circulating transthyretin (TTR) [9]. It affects around 25 % of patients greater than 80 years of age and is much more common in males. The median OS from diagnosis of CHF is 75 months, compared to less than a year for AL amyloidosis [10].
Familial/Hereditary Systemic Amyloidosis (Mutant ATTR)
The most common cause of hereditary amyloidosis is a mutant ATTR protein, rendering it amyloidogenic. Val30Met is the most common mutation globally [11]. In black Americans older than 50 years of age, a point mutation leading to a valine-to-isoleucine substitution (V122I) was found in 4 % of individuals, and was associated with an increased risk of incident systolic and diastolic heart failure [12]. However, the overall mortality in carriers of this mutation was not increased compared to non-carriers and manifestations of overt cardiomyopathy was low (7 %). The prevalence of mutant ATTR amyloidosis in the USA is estimated to be 0.4 per million/year [13]. Neuropathy and cardiomyopathy are the cardinal manifestations of mutant ATTR amyloidosis. The 2-year OS rates in mutant ATTR and wt ATTR subtypes are similar [14].
Secondary Systemic Amyloidosis (AA)
Secondary amyloidosis results from excessive production of the acute phase reactant serum amyloid A (SAA) protein, due to chronic inflammation of any source. In the western world, it is usually associated with autoimmune disorders. The kidney is the most common organ involved [15], with cardiac involvement being rare.
Isolated Atrial Amyloidosis (IAA)
IAA or atrial natriuretic factor-related amyloidosis (AANF) is characterized by the deposition of atrial natriuretic peptide in the hearts of elderly patients [16]. It is a localized form of amyloidosis and is usually identified on autopsy examination. AANF is more common in women and is clinically associated with an increased risk of atrial tachyarrhythmia [4].
Management
Basic Principles of Management
The basic foundation for the management of cardiac amyloidosis rests on two pillars: treatment of the underlying condition to suppress the production of the amyloidogenic protein and management of heart failure-related symptoms [4]. Amyloid cardiomyopathy is best managed by a multidisciplinary group of specialists in a tertiary care center, which includes hematologists, cardiologists, and nephrologists.
Supportive Care
Diuresis, oral or intravenous, remains the mainstay of heart-failure treatment, with escalating doses often required due to concomitant hypoalbuminemia in the setting of nephrotic syndrome. There is inadequate data on the efficacy of angiotensin-converting enzymes (ACE) inhibitors, angiotensin receptor blockers (ARBs), and beta-blockers in the treatment of cardiac amyloidosis [4]. Furthermore, use of ACE inhibitors is limited by significant hypotension even at low doses, due to reduction in cardiac preload. Beta blockers may produce bradycardia and hypotension and are poorly tolerated. Centrally acting calcium channel blockers should be avoided, due to the risk of worsening CHF, likely as a result of their negative inotropic effect [17]. Digoxin should be used with caution, as it can bind with amyloid fibrils, leading to locally high drug concentrations [18].
Role of Cardiac Implantable Devices
Sudden cardiac death due to arrhythmias is an important concern in amyloid heart disease and warrants consideration for defibrillator placement. A study on 19 patients with biopsy-proven cardiac amyloidosis and at high-risk for arrhythmias who underwent placement of implantable cardioverter defibrillator (ICD) showed the predominant cause of death to be electromechanical dissociation (n = 6), which was not amenable to defibrillation [19]. Two patients developed sustained ventricular tachyarrhythmia and were successfully treated by the ICD. A single-institution study on ICD placement in cardiac amyloidosis showed the indication to be primary prevention in 77 % and secondary prevention in 23 % of the cases [20]. The rate of appropriate shocks in the first year of placement was 32 % and was seen exclusively in those with AL cardiac amyloidosis. In patients with familial amyloidosis, prophylactic pacemaker implantation prevented major cardiac events in 25 % of patients over a 4-year follow-up.
Placement of a left ventricular assist device (LVAD) as a bridge to heart transplantation in patients with amyloid cardiomyopathy is technically viable, with 4 out of 9 implanted patients alive at a follow-up of 16–24 months in a study [21]. The 1-year OS rate without transplantation post-LVAD implant is 64 %, which is similar to that seen in non-amyloid cardiomyopathy [22].
Treatment directed to suppression of amyloid protein production by amyloidosis subtypes will be discussed below and has been summarized in Table 2.
AL Amyloidosis
The treatment of AL amyloidosis involves destruction of the plasma cell clone responsible for production of the amyloidogenic light chain. Systemic chemotherapy has been shown to improve organ function, OS, and quality of life (QoL) [15]. FLC response, defined as the percentage decrease in difference between involved and uninvolved FLC (FLC-diff), was shown to be a stronger predictor of OS compared to M-protein response in AL amyloidosis [23]. Very good partial response (VGPR) after autologous stem cell transplantation (ASCT) in patients with AL cardiac amyloidosis translates into superior OS [24], with 5-year OS rates in patients achieving ≥90 % decrease in FLC-diff with treatment exceeding 70 % [23]. Among a poor prognostic subgroup of Mayo stage III patients with NT-Pro-BNP > 8500 ng/ml, those achieving CR or VGPR have median OS of 28 months, compared to 2.6 months in those with no response [25]. From the organ response standpoint, cardiac response is defined as a decrease in NT-proBNP by >30 % and >300 pg/ml (if baseline NT-proBNP >650 pg/ml), or a ≥2-point decrease in NYHA class (if baseline NYHA class III or IV) [26].
Alkylator-Based Treatment and ASCT
An alkylator, in combination with a corticosteroid, has been widely used as a first-line therapy in AL amyloidosis. A randomized controlled trial (RCT) of chemotherapy regimens in AL amyloidosis showed melphalan and prednisone to be superior to colchicine monotherapy [27]. Oral melphalan and dexamethasone (MelDex) in patients ineligible for ASCT leads to an overall response rate (ORR) close to 70 %, with a median PFS and OS of 3.8 and 5.1 years, respectively [28]. However, results in patients with advanced cardiac involvement are still poor with MelDex. A RCT comparing high-dose melphalan followed by ASCT (HDM/ASCT) and MelDex in AL amyloidosis found significantly longer median OS with the latter (22.2 with HDM/ASCT vs 56.9 months with MelDex, respectively; p = 0.04) [29]. However, transplant-related mortality (TRM) in this study was exceptionally high at 24 % and 10 out of 37 patients in the HDM/ASCT arm were administered reduced dose melphalan, which has been shown to be inferior to standard dose melphalan in amyloidosis [30, 31]. Furthermore, patients with severe cardiac involvement did poorly irrespective of the treatment arm with a 3-year OS rate of 25 %.
HDM/ASCT leads to a durable remission and an impressive median OS of 10 years in those achieving a CR after transplant [30]. In patients with cardiac amyloidosis achieving a CR after HDM/ASCT, 10-year OS rate is around 60 % [24]. A study from the Center for International Blood and Marrow Transplant Research (CIBMTR) registry showed improving outcomes with HDM/ASCT in recent years, with 100-day mortality rate after ASCT decreasing from 20 % (1995–2000 cohort) to 5 % (2007–2012 cohort) [31]. Five-year OS rates have improved from 55 to 77 % during the same time period. However, in patients with cardiac amyloidosis, there was only a modest improvement in 3-year OS rate (62 % in 2001–2006 vs 67 % in 2007–2012 cohort; p = 0.59), due to a high rate of early mortality after transplant. Therefore, a risk-adapted approach should be used to select patients with less severe cardiac involvement for HDM/ASCT. The eligibility criteria for HDM/ASCT in Mayo Clinic are summarized in Table 3.
Proteasome Inhibitors
Novel agents, including proteasome inhibitors (PIs) and immunomodulators (IMiDs), have shown excellent activity in both frontline and relapsed setting in AL amyloidosis. A study of bortezomib with or without dexamethasone showed a hematologic CR rate of 47 and 25 % in newly diagnosed (ND) and relapsed/refractory (R/R) disease, respectively, with a 1-year OS rate of 76 % [32]. A cardiac response was seen in 30 % of patients, mostly manifesting as an improvement in their NYHA functional class, and was closely associated with hematologic response. A phase 1/2 study comparing once weekly and twice weekly bortezomib in relapsed AL amyloidosis showed fewer serious adverse events (AEs) in the former (50 vs 79 %, respectively) [33, 34].
Cyclophosphamide, bortezomib, and dexamethasone (CyBorD) has been shown to produce deep responses, with a CR rate of 65 and 22 % in ND and R/R patients, respectively [35]. The regimen was well tolerated in patients with cardiac involvement, which included 74 % of the study cohort. The 2-year OS rate in patients with Mayo stage III was 94 % with CyBorD. As frontline treatment, CyBorD leads to an ORR and very good partial response (VGPR) rate of 62 and 43 %, respectively, with cardiac response in 17 % of patients. However, Mayo stage III patients had a lower VGPR rate of 23 % and 2-year OS rate of 67 % in responders [36].
Immunomodulators
Lenalidomide, in combination with MelDex, was shown to be active in ND AL amyloidosis, with a CR rate of 42 % and estimated 2-year PFS and OS rates of 54 and 81 %, respectively, at a dose of 15 mg/day [37]. Lenalidomide and dexamethasone (Rd) was subsequently shown to have promising activity in both ND and R/R AL amyloidosis, with an ORR of 43–67 % and a CR rate of 29 % [38, 39]. In R/R patients with prior exposure to thalidomide and/or bortezomib, Rd led to a CR rate of 20 %, with 2-year PFS and OS rates of 73 and 84 %, respectively. Fifty percent of patients had cardiac involvement and 12 % had achieved a cardiac response with Rd [40]. Cyclophosphamide, lenalidomide, and dexamethasone (CRd) led to an ORR of 60 %, with 40 % being VGPR or better, and a cardiac response rate of 23 % [41]. The 2-year OS rates in patients completing at least 3 cycles of treatment was 70 %, which was comparable to that seen with Rd alone. Notably, early death during treatment with CRd was seen in 20 % of patients, all of whom had cardiac involvement.
Pomalidomide has shown activity in R/R AL amyloidosis, with an ORR of 48 % and 1-year PFS and OS rates of 59 and 76 %, respectively. Eighty-two percent had cardiac amyloidosis and 25 % had Mayo cardiac stage III disease in this study [42]. However, an important caveat to be noted while treating these patients with IMiDs is that NT-pro-BNP can rise during treatment despite hematologic response [42, 43]. Although the reason for this laboratory abnormality is unclear, rise in NT-pro BNP despite hematological response can potentially implicate poorer prognosis and should not be disregarded.
Monoclonal Antibodies
All amyloidogenic proteins contain a backbone of normal plasma glycoprotein, called serum amyloid P (SAP). Anti-human SAP monoclonal antibodies have been demonstrated in mice to mediate a complement-dependent immunological reaction, which clears visceral amyloid deposits [44]. Following depletion of circulating human SAP by bis-d-proline compound CPHPC [45], amyloid deposits can be cleared by treatment with SAP-binding monoclonal antibody. Based on these pre-clinical studies, a phase 1 trial was performed in 15 patients with systemic amyloidosis, in which humanized monoclonal IgG1 anti-SAP was administered after CPHPC administration [46]. A reduction of hepatic and renal amyloid load, as measured by SAP scintigraphy, was noted. However, due to potential immune-mediated cardiotoxicity, patients with cardiac amyloidosis were excluded from this study. Another monoclonal antibody named NEOD001 targeting the misfolded light chains in AL amyloidosis, was studied in patients with refractory AL amyloidosis and persistent organ dysfunction [47]. Cardiac response and stable cardiac disease was noted in 57 and 43 % of evaluable patients, respectively, which compares favorably to conventional chemotherapy regimens. No grade 3–4 AEs were noted with this drug. A phase 1 study of monoclonal antibody 11-1F4 against human light chain related fibrils showed a cardiac response in two patients, with no serious AEs [48].
Doxycycline
Doxycycline has been shown to reduce light chain fibril formation in transgenic mouse models of AL amyloidosis [49]. Ex vivo treatment of light chain fibrils with doxycycline leads to a reduced number of intact fibrils. A matched case–control study of CyBorD with or without doxycycline has shown to significantly reduce early mortality, higher CR/VGPR rate, higher cardiac responses and superior 2-year OS rate (82 vs 40 %) with doxycycline [50, 51]. However, prospective studies are needed before making firm conclusions.
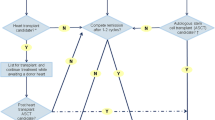
Role of Orthotopic Heart Transplantation (OHT) and Combined Heart/Stem Cell Transplantation
OHT alone has been reported in AL cardiac amyloidosis, but early enthusiasm was tempered by poor long-term survival, likely due to persistent production of the pathogenic amyloid protein [15]. A series of 17 patients undergoing OHT showed a higher 5-year OS rate in patients who had received prior chemotherapy, compared to those who had not (36 vs 20 %, respectively) [52]. Amyloid recurrence and mortality due to extra-cardiac manifestations was significantly higher in those previously untreated patients. Long-term survival in transplanted patients with amyloid heart disease was shown to be significantly inferior compared to those receiving cardiac transplant for other indications [53].
Use of ASCT after OHT to suppress the plasma cell clone producing the amyloidogenic light chain has led to better outcomes. A single-center study on 11 patients undergoing heart transplantation, followed by HDM/ASCT, showed a 5-year OS rate of 65 %. TRM was 18 % and 3 patients died of progressive amyloidosis at 55–66 months after ASCT [54]. Another study on 9 patients with AL cardiac amyloidosis, 5 of whom had undergone prior ASCT, showed a 1-year post-transplant OS rate of 100 % [55]. Since TRM with ASCT is high in patients with cardiac amyloidosis, prospective studies are required to ascertain the risk-benefit ratio in this population.
Transthyretin Amyloidosis (ATTR Amyloidosis)
Therapies for ATTR amyloidosis include liver transplantation to suppress the production of mutant TTR by the liver and pharmacotherapy [13]. Combined liver and cardiac transplantation, as well as isolated cardiac transplantation, has also been studied in these patients with promising results.
Role of Orthotopic Liver Transplantation (OLT)
The therapeutic rationale for OLT in hereditary systemic amyloidosis is replacement of the mutant TTR, predominantly produced in the liver. However, deposition of both mutant and wt TTR in cardiac tissue was found at autopsy of patients with hereditary amyloidosis undergoing OLT [56]. Furthermore, in a study involving more than 10 years of follow-up of patients with hereditary amyloidosis undergoing OLT, moderate to severe wt TTR deposits were noted in the heart, tongue, and spinal cord [57]. This indicated a shift from mutant to wt TTR deposition, produced by normal liver grafts in such patients. The ratio of mutant to wt TTR deposition in cardiac tissues of patients undergoing OLT was 25:75, compared to a ratio of 60:40 in those not undergoing OLT. An increase in the thickness of the interventricular septum was also noted in patients after OLT, especially in males with late onset of mutant TTR amyloidosis [58].
Long-term data on outcomes of OLT in hereditary amyloidosis showed 5-year OS rates of 82–100 and 59 % in patients with Val30Met and non-Val30Met TTR variants, respectively [59, 60]. Cardiac-related causes were responsible for death in 39 % of patients. Survival in patients with the Val30Met mutation had improved over time with OLT, with 5-year OS rates increasing from 60 % in 1990–1994 to 90 % in the 2005–2009 periods [59]. Lower survival after OLT is related to malnutrition, later onset and longer duration of disease and presence of cardiomyopathy at transplant. The quality of life (QoL) at a follow-up of 4 years or more post-transplantation was worse in patients with hereditary amyloidosis, compared to those undergoing OLT for other indications [61], which could be related to ongoing wt TTR deposition.
Role of Cardiac and Combined Cardiac/Liver Transplantation
The rationale behind the combination of cardiac and liver transplantation is removal of the mutant TTR nidus deposited in cardiac tissues, which serves as the backbone for subsequent wt TTR deposition, along with eliminating the production of mutant TTR by the liver. Combined cardiac and liver transplantation has yielded superior survival outcomes, compared to OLT alone. A study on long-term outcomes of patient with leu111Met TTR mutation undergoing combined liver-heart transplantation showed an OS rate of 71 % at 4.5 years [62]. All 7 patients undergoing transplant had severe cardiomyopathy, with a median left ventricular EF of 40 % at transplant. Among 5 surviving patients, left ventricular EF was 61 ± 2 %, with no recurrence of cardiac amyloid noted after transplant, at a median follow-up of 55 months. In a study on 25 patients with mutant TTR amyloidosis, 5 successfully underwent cardiac transplantation and 1 patient subsequently had an OLT [63]. Four out of 5 patients did not have amyloid deposition in cardiac tissue, by EMB, after transplant.
A case of isolated heart transplantation has been reported in a patient with heterozygous V122I mutated TTR cardiac amyloidosis [64]. The patient had no recurrence of cardiac amyloid 5 years after transplantation. A series of 10 patients with TTR cardiac amyloidosis undergoing heart transplant showed recurrence of amyloid in the cardiac allograft in only 1 patient, with a 1-year OS rate of 74 % [55].
Pharmacologic therapy
Since liver and cardiac transplantation is not feasible in most patients with ATTR amyloidosis, especially in the elderly, effective pharmacologic therapy is needed. Diflunisal, a non-steroidal anti-inflammatory drug, has been shown to bind with and stabilize TTR tetramers and prevent deposition of misfolded protein in tissues [65]. In a study of diflunisal in ATTR cardiac amyloidosis (both mutant and wt), stable mean left ventricular (LV) mass, LVEF and cardiac biomarkers were seen during the course of therapy [66]. Nephrotoxicity and volume overload was observed as AEs. A RCT of diflunisal for familial amyloid polyneuropathy (FAP; caused by mutant ATTR) reported neurological stability at 2 years in 29.7 % of patients in the diflunisal arm versus 9.4 % of the placebo arm [67].
Tafamidis, a kinetic stabilizer of TTR protein, is active in FAP and leads to a delay in peripheral neurologic impairment, with minimal AEs [68, 69]. A phase 2 study of tafamidis in ATTR amyloid cardiomyopathy showed TTR stabilization in 97 % of wt patients with mild to moderate CHF [70]. There were no clinically significant changes in cardiac biomarkers or echocardiography on follow-up. In patients with non-Val30Met TTR variants, tafamidis leads to stabilization of the amyloid protein and stable QoL, cardiac biomarkers, echocardiographic parameters, and body mass index during the course of treatment [71].
Small interfering ribonucleic acid (siRNA), which can selectively suppress TTR gene expression in hepatocytes, has been tested in formulations of lipid nanoparticles in a phase 1 study [72]. Suppression of both wt and mutant TTR protein was seen, with the only significant AE being infusion site reactions. Antisense oligonucleotides, complementary to the messenger RNA molecule encoding TTR in hepatocytes mediate destruction of the mRNA by ribonuclease-H upon binding, thereby reducing translation and suppressing levels of serum TTR [73, 74].
Doxycycline was shown to disrupt TTR amyloid fibrils in vitro in preclinical studies [75]. A phase 2 study of doxycycline and tauroursodeoxycholic acid in ATTR amyloidosis (both mutant and wt) showed no progression of cardiomyopathy during the course of treatment [76]. No serious AEs were noted. Green tea and green tea extracts, containing Epigallocatechin-3-gallate, has been shown to prevent progression of ATTR amyloidosis in an observational study [77].
Secondary systemic amyloidosis (AA)
Cardiac involvement is rare in AA amyloidosis, with renal dysfunction being the main prognostic marker which predicts OS [78]. In case of cardiac involvement, symptomatic management of heart failure symptoms as described above, along with therapy directed to suppress underlying chronic inflammatory condition is the key to management.
Conclusion
Early diagnosis, risk-adapted approach, and plasma cell directed therapy have led to improved outcomes in AL cardiac amyloidosis. However, patients with advanced disease and those who do not achieve a deep hematologic response to therapy still have poor outcomes. A decrease in TRM with HDM/ASCT in AL amyloidosis in recent years and the introduction of the novel agents, including PIs and IMiDs, are promising and warrants prospective randomized studies comparing transplant and non-transplant strategies. Monoclonal antibodies directed against the amyloid fibrils have shown cardiac response in phase 1/2 studies. OLT remains the benchmark for treatment of ATTR amyloidosis; however, progression of cardiac amyloidosis after OLT due to an increase in the wild:mutant TTR ratio is concerning. Development of “TTR stabilizers” is a promising arena in hereditary amyloidosis and further studies are needed to ascertain survival benefit in those with cardiac involvement.
References
Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349(6):583–96.
Gertz MA, Comenzo R, Falk RH, Fermand JP, Hazenberg BP, Hawkins PN, et al. Definition of organ involvement and treatment response in immunoglobulin light chain amyloidosis (AL): a consensus opinion from the 10th International Symposium on Amyloid and Amyloidosis, Tours, France, 18–22 April 2004. Am J Hematol. 2005;79(4):319–28.
Smith TJ, Kyle RA, Lie JT. Clinical significance of histopathologic patterns of cardiac amyloidosis. Mayo Clin Proc. 1984;59(8):547–55.
Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112(13):2047–60.
Kyle RA, Linos A, Beard CM, Linke RP, Gertz MA, O’Fallon WM, et al. Incidence and natural history of primary systemic amyloidosis in Olmsted County, Minnesota, 1950 through 1989. Blood. 1992;79(7):1817–22.
Pinney JH, Smith CJ, Taube JB, Lachmann HJ, Venner CP, Gibbs SD, et al. Systemic amyloidosis in England: an epidemiological study. Br J Haematol. 2013;161(4):525–32.
Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Colby C, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012;30(9):989–95.
Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, et al. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91(2):141–57.
Westermark P, Sletten K, Johansson B, Cornwell 3rd GG. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87(7):2843–5.
Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med. 2005;165(12):1425–9.
Ohmori H, Ando Y, Makita Y, Onouchi Y, Nakajima T, Saraiva MJ, et al. Common origin of the Val30Met mutation responsible for the amyloidogenic transthyretin type of familial amyloidotic polyneuropathy. J Med Genet. 2004;41(4):e51.
Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–9.
Gertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, Cruz M, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015;66(21):2451–66.
Rapezzi C, Merlini G, Quarta CC, Riva L, Longhi S, Leone O, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–12.
Sher T, Gertz MA. Recent advances in the diagnosis and management of cardiac amyloidosis. Futur Cardiol. 2014;10(1):131–46.
Pucci A, Wharton J, Arbustini E, Grasso M, Diegoli M, Needleman P, et al. Atrial amyloid deposits in the failing human heart display both atrial and brain natriuretic peptide-like immunoreactivity. J Pathol. 1991;165(3):235–41.
Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55(13 Pt 1):1645.
Cassidy JT. Cardiac amyloidosis. Two cases with digitalis sensitivity. Ann Intern Med. 1961;55:989–94.
Kristen AV, Dengler TJ, Hegenbart U, Schonland SO, Goldschmidt H, Sack FU, et al. Prophylactic implantation of cardioverter-defibrillator in patients with severe cardiac amyloidosis and high risk for sudden cardiac death. Heart rhythm. 2008;5(2):235–40.
Lin G, Dispenzieri A, Kyle R, Grogan M, Brady PA. Implantable cardioverter defibrillators in patients with cardiac amyloidosis. J Cardiovasc Electrophysiol. 2013;24(7):793–8.
Swiecicki PL, Edwards BS, Kushwaha SS, Dispenzieri A, Park SJ, Gertz MA. Left ventricular device implantation for advanced cardiac amyloidosis. J Heart Lung Transplant. 2013;32(5):563–8.
Grupper A, Park SJ, Pereira NL, Schettle SD, Gerber Y, Topilsky Y, et al. Role of ventricular assist therapy for patients with heart failure and restrictive physiology: Improving outcomes for a lethal disease. J Heart Lung Transplant. 2015;34(8):1042–9.
Kumar SK, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, Zeldenrust SR, et al. Changes in serum-free light chain rather than intact monoclonal immunoglobulin levels predicts outcome following therapy in primary amyloidosis. Am J Hematol. 2011;86(3):251–5.
Gertz MA, Lacy MQ, Dispenzieri A, Hayman SR, Kumar SK, Leung N, et al. Effect of hematologic response on outcome of patients undergoing transplantation for primary amyloidosis: importance of achieving a complete response. Haematologica. 2007;92(10):1415–8.
Sevillano B, Wechalekar A, Foard D, Whelan C, Fontana M, Quarta C, et al. A prospective study of treatment outcomes in 179 patients with advanced cardiac stage STAGE IIIb amyloidosis. Blood. 2015;126(23):5357.
Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A, Hawkins PN, et al. New criteria for response to treatment in immunoglobulin light chain amyloidosis based on free light chain measurement and cardiac biomarkers: impact on survival outcomes. J Clin Oncol. 2012;30(36):4541–9.
Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, et al. A trial of three regimens for primary amyloidosis: colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N Engl J Med. 1997;336(17):1202–7.
Palladini G, Russo P, Nuvolone M, Lavatelli F, Perfetti V, Obici L, et al. Treatment with oral melphalan plus dexamethasone produces long-term remissions in AL amyloidosis. Blood. 2007;110(2):787–8.
Jaccard A, Moreau P, Leblond V, Leleu X, Benboubker L, Hermine O, et al. High-dose melphalan versus melphalan plus dexamethasone for AL amyloidosis. N Engl J Med. 2007;357(11):1083–93.
Sanchorawala V, Skinner M, Quillen K, Finn KT, Doros G, Seldin DC. Long-term outcome of patients with AL amyloidosis treated with high-dose melphalan and stem-cell transplantation. Blood. 2007;110(10):3561–3.
D’Souza A, Dispenzieri A, Wirk B, Zhang MJ, Huang J, Gertz MA, et al. Improved outcomes after autologous hematopoietic cell transplantation for light chain amyloidosis: a center for international blood and marrow transplant research study. J clin Oncol. 2015;33(32):3741–9.
Kastritis E, Wechalekar AD, Dimopoulos MA, Merlini G, Hawkins PN, Perfetti V, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol. 2010;28(6):1031–7.
Reece DE, Hegenbart U, Sanchorawala V, Merlini G, Palladini G, Blade J, et al. Efficacy and safety of once-weekly and twice-weekly bortezomib in patients with relapsed systemic AL amyloidosis: results of a phase 1/2 study. Blood. 2011;118(4):865–73.
Dubrey SW, Reece DE, Sanchorawala V, Hegenbart U, Merlini G, Palladini G, et al. Bortezomib in a phase 1 trial for patients with relapsed AL amyloidosis: cardiac responses and overall effects. QJM. 2011;104(11):957–70.
Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH, et al. Cyclophosphamide, bortezomib, and dexamethasone therapy in AL amyloidosis is associated with high clonal response rates and prolonged progression-free survival. Blood. 2012;119(19):4387–90.
Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612–5.
Moreau P, Jaccard A, Benboubker L, Royer B, Leleu X, Bridoux F, et al. Lenalidomide in combination with melphalan and dexamethasone in patients with newly diagnosed AL amyloidosis: a multicenter phase 1/2 dose-escalation study. Blood. 2010;116(23):4777–82.
Sanchorawala V, Wright DG, Rosenzweig M, Finn KT, Fennessey S, Zeldis JB, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood. 2007;109(2):492–6.
Dispenzieri A, Lacy MQ, Zeldenrust SR, Hayman SR, Kumar SK, Geyer SM, et al. The activity of lenalidomide with or without dexamethasone in patients with primary systemic amyloidosis. Blood. 2007;109(2):465–70.
Mahmood S, Venner CP, Sachchithanantham S, Lane T, Rannigan L, Foard D, et al. Lenalidomide and dexamethasone for systemic AL amyloidosis following prior treatment with thalidomide or bortezomib regimens. Br J Haematol. 2014;166(6):842–8.
Kumar SK, Hayman SR, Buadi FK, Roy V, Lacy MQ, Gertz MA, et al. Lenalidomide, cyclophosphamide, and dexamethasone (CRd) for light-chain amyloidosis: long-term results from a phase 2 trial. Blood. 2012;119(21):4860–7.
Dispenzieri A, Buadi F, Laumann K, Laplant B, Hayman SR, Kumar SK, et al. Activity of pomalidomide in patients with immunoglobulin light-chain amyloidosis. Blood. 2012;119(23):5397–404.
Dispenzieri A, Dingli D, Kumar SK, Rajkumar SV, Lacy MQ, Hayman S, et al. Discordance between serum cardiac biomarker and immunoglobulin-free light-chain response in patients with immunoglobulin light-chain amyloidosis treated with immune modulatory drugs. Am J Hematol. 2010;85(10):757–9.
Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–7.
Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417(6886):254–9.
Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373(12):1106–14.
Gertz MA, Landau H, Comenzo RL, Seldin D, Weiss B, Zonder J, et al. First-in-human phase I/II study of NEOD001 in patients with light chain amyloidosis and persistent organ dysfunction. J Clin Oncol. 2016;34(10):1097–103.
Langer AL, Miao S, Mapara M, Radhakrishnan J, Maurer MS, Raza S, et al. Results of phase I study of chimeric fibril-reactive monoclonal antibody 11-1F4 in patients with AL amyloidosis. Blood. 2015;126(23):188.
Ward JE, Ren R, Toraldo G, Soohoo P, Guan J, O’Hara C, et al. Doxycycline reduces fibril formation in a transgenic mouse model of AL amyloidosis. Blood. 2011;118(25):6610–7.
Wechalekar A, Whelan C, Sachchithanantham S, Fontana M, Mahmood S, Foard D, et al. A matched case control study of doxycycline added to chemotherapy for reducing early mortality in patients with advanced cardiac AL amyloidosis from the alchemy study cohort. Blood. 2014;124(21):3485.
Wechalekar A, Whelan C, Lachmann H, Fontana M, Mahmood S, Gillmore JD, et al. Oral doxycycline improves outcomes of stage III AL amyloidosis—a matched case control study. Blood. 2015;126(23):732.
Dubrey SW, Burke MM, Hawkins PN, Banner NR. Cardiac transplantation for amyloid heart disease: the United Kingdom experience. J Heart Lung Transplant. 2004;23(10):1142–53.
Hosenpud JD, Demarco T, Frazier OH, Griffith BP, Uretsky BF, Menkis AH, et al. Progression of systemic disease and reduced long-term survival in patients with cardiac amyloidosis undergoing heart transplantation. Follow-up results of a multicenter survey. Circulation. 1991;84(5):338–43.
Lacy MQ, Dispenzieri A, Hayman SR, Kumar S, Kyle RA, Rajkumar SV, et al. Autologous stem cell transplant after heart transplant for light chain (Al) amyloid cardiomyopathy. J Heart Lung Transplant. 2008;27(8):823–9.
Davis MK, Kale P, Liedtke M, Schrier S, Arai S, Wheeler M, et al. Outcomes after heart transplantation for amyloid cardiomyopathy in the modern era. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surg. 2015;15(3):650–8.
Tasaki M, Ueda M, Obayashi K, Koike H, Kitagawa K, Ogi Y, et al. Effect of age and sex differences on wild-type transthyretin amyloid formation in familial amyloidotic polyneuropathy: a proteomic approach. Int J Cardiol. 2013;170(1):69–74.
Oshima T, Kawahara S, Ueda M, Kawakami Y, Tanaka R, Okazaki T, et al. Changes in pathological and biochemical findings of systemic tissue sites in familial amyloid polyneuropathy more than 10 years after liver transplantation. J Neurol Neurosurg Psychiatry. 2014;85(7):740–6.
Okamoto S, Zhao Y, Lindqvist P, Backman C, Ericzon BG, Wijayatunga P, et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary transthyretin amyloidosis (ATTR) patients. Amyloid. 2011;18(4):200–5.
Wilczek HE, Larsson M, Ericzon BG. Long-term data from the familial amyloidotic polyneuropathy world transplant registry (FAPWTR). Amyloid. 2011;18 Suppl 1:193–5.
Carvalho A, Rocha A, Lobato L. Liver transplantation in transthyretin amyloidosis: issues and challenges. Liver Transplant. 2015;21(3):282–92.
Drent G, Graveland CW, Hazenberg BP, Haagsma EB. Quality of life in patients with familial amyloidotic polyneuropathy long-term after liver transplantation. Amyloid. 2009;16(3):133–41.
Nelson LM, Penninga L, Sander K, Hansen PB, Villadsen GE, Rasmussen A, et al. Long-term outcome in patients treated with combined heart and liver transplantation for familial amyloidotic cardiomyopathy. Clin Transpl. 2013;27(2):203–9.
Sack FU, Kristen A, Goldschmidt H, Schnabel PA, Dengler T, Koch A, et al. Treatment options for severe cardiac amyloidosis: heart transplantation combined with chemotherapy and stem cell transplantation for patients with AL-amyloidosis and heart and liver transplantation for patients with ATTR-amyloidosis. European J Cardiothoracic Surg. 2008;33(2):257–62.
Thenappan T, Fedson S, Rich J, Murks C, Husain A, Pogoriler J, et al. Isolated heart transplantation for familial transthyretin (TTR) V122I cardiac amyloidosis. Amyloid. 2014;21(2):120–3.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–49.
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315–9.
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. Jama. 2013;310(24):2658–67.
Coelho T, Maia LF, Martins Da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92.
Coelho T, Maia LF, da Silva AM, Cruz MW, Plante-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–14.
Maurer MS, Grogan DR, Judge DP, Mundayat R, Packman J, Lombardo I, et al. Tafamidis in transthyretin amyloid cardiomyopathy: effects on transthyretin stabilization and clinical outcomes. Circ Heart Fail. 2015;8(3):519–26.
Merlini G, Plante-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011–20.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29.
Benson MD, Pandey S, Witchell D, Jazayeri A, Siwkowski A, Monia B, et al. Antisense oligonucleotide therapy for TTR amyloidosis. Amyloid. 2011;1(18):60.
Ackermann EJ, Guo S, Booten S, Alvarado L, Benson M, Hughes S, et al. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19 Suppl 1:43–4.
Cardoso I, Merlini G, Saraiva MJ. 4′-iodo-4′-deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: screening for TTR fibril disrupters. FASEB J. 2003;17(8):803–9.
Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19(1):34–6.
Kristen AV, Lehrke S, Buss S, Mereles D, Steen H, Ehlermann P, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol. 2012;101(10):805–13.
Lachmann HJ, Goodman HJ, Gilbertson JA, Gallimore JR, Sabin CA, Gillmore JD, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356(23):2361–71.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Rajshekhar Chakraborty and Eli Muchtar declare that they have no conflict of interest.
Morie A. Gertz has received personal fees from Prothena, Ionis, GSK, and Annexon during the conduct of the study.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Management of Heart Failure
Rights and permissions
About this article

Cite this article
Chakraborty, R., Muchtar, E. & Gertz, M.A. Newer Therapies for Amyloid Cardiomyopathy. Curr Heart Fail Rep 13, 237–246 (2016). https://doi.org/10.1007/s11897-016-0300-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-016-0300-1