
Abstract
IL-33, a member of the IL-1 cytokine family and a ligand to receptor ST2, has great potential to induce a T helper 2-type inflammatory response. IL-33 is proven to be released by epithelial cells during their injury by different environmental stimuli such as airborne allergens, viruses, and air pollutants. IL-33 acting as an endogenous danger signal is termed an alarmin. As such, this cytokine is considered to play a crucial role in an allergic inflammatory disease such as rhinitis. Recent investigations regarding the IL-33/ST2 axis involvement in Th2 inflammatory response and pathogenesis of rhinitis have been reviewed. The role of IL-33 as a novel promising therapeutic target has also been discussed.
Similar content being viewed by others


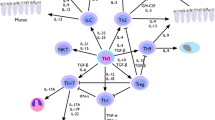
Introduction
IL-33 is a member of the IL-1 family of cytokines, such as IL-1β and IL-18. IL-33 is also known as a member of mesenchymal-derived cytokines such as IL-25, thymic stromal lymphopoietin (TSLP). IL-33 is an interesting cytokine because of it has either proinflammatory or anti-inflammatory properties. IL-33 is believed to be involved in Th2-mediated inflammatory responses in allergic diseases such as asthma, anaphylaxis, and atopic dermatitis, and in host defense against parasites. On the other hand, IL-33 has a protective effect in atherosclerosis, obesity, type 2 diabetes, and cardiac remodeling [1]. Moreover, IL-33 is thought to be a bridge between innate and acquire immune defences [2•].
IL-33 was originally identified as “DVS27” and as a nuclear factor protein in high endothelial venules; thus, it was called NF-HEV [3, 4]. As a result of searching for the sequences containing a β-trefoil structure, it has been termed as IL-33 and qualifies as a member of the IL-1 cytokine family [5]. The first report describing this protein as IL-33 came from 2005; that is why our understanding of the role and function of this cytokine is still evolving [5].
Stimulation of IL-33 Release and its Activation
IL-33 may be released by epithelial cells during their injury or necrosis caused by exogenous triggers such as mechanical trauma, viruses, smoke, airborne allergens, or endogenous triggers. These triggers may also lead to activation of pattern recognition receptors such as TLRs, and as a consequence, the release of IL-33 from epithelial cells [2•]. The fact that necrotic cells release uncleaved but biologically active IL-33 [2•] suggests that IL-33 may act as a endogenous danger signal; thus, it has been termed an alarmin [2•, 6, 7].
Similarly to IL-1β and IL-18, IL-33 is proteolytically cleaved by caspases. However, the former cytokines are activated in inflammasome during apoptosis and their cleaved forms become biologically active and secreted [8]. In contrast, full-length IL-33 passively secreted by necrotic cells is biologically active in vivo and proteolysis by caspase-1 from apoptotic cells suppresses or even inactivates IL-33 bioactivity [9–11, 12•]. Thus, apoptotic cells inactivate IL-33 and prevent the development of type 2 immune responses.
Receptor for IL-33
IL-33 is a ligand for a heterodimeric membrane-bound receptor consisting of the orphan IL-1 family receptor ST2 (suppression of tumorigenecity 2), also called ST2L, T1, Der-4 and fit-1, and the ubiquitously expressed IL-1 receptor-accessory protein IL-1RAcP, whereas ST2 expression is restricted to IL-33 stimulation [5, 13].
IL-33 acts through activation of the ST2L receptor complex, but also as an intracellular nuclear factor with transcriptional regulatory/repressing properties [14, 15]. There are at least three isoforms of ST2 gene production, resulting from different splicing of the gene, a soluble form of ST2 (sST2), a membrane-bound form (ST2L), and a constitutively active variant ST2 (ST2V) [2•, 16, 17]. Soluble ST2 acts as a decoy receptor for IL-33 and functionally inhibits IL-33 activity in vitro and in vivo [18].
Many cells express ST2L receptor for IL-33. ST2L is expressed on the earliest human progenitors cells such as CD34+ cells and on the multiple stages of development of leukocytes. Moderate levels of ST2L are expressed on resting eosinophils, which increased in the airways of mice with OVA- induced asthma. ST2 is expressed on Th2 cells, but not Th1 cells [19–21]. Similarly, ST2 is expressed on in vitro skewed Tc2 cells rather than Tc1 cells [22]. The expression of ST2 was also found in mast cells, basophils, macrophages, human and mouse iNKT cells, NK cells and B-1 cells, but not in neutrophils [2•, 23, 24].
Elevated serum level of ST2 was found in adults and children with acute exacerbation of asthma but not in seasonal allergic rhinitis patients [25–27]. However, expression of ST2 was significantly higher in the nasal epithelium in allergic rhinitis patients [28].
Cellular Source of IL-33
IL-33 is constitutively expressed on plenty of cells and multiple tissues, among them by epithelial barrier tissues, such as nasal, bronchial or ocular epithelial cells, human and mouse mast cells derived from peripheral blood or cord blood progenitor cells, lymphoid organs, brain, embryos, inflamed tissues, smooth muscle cells, keratinocytes, macrophages and dendritic cells, and others [28–32].
Activity of IL-33 in Cells Participating in Allergic Inflammation
IL-33 has a plenipotent and pleiotropic activity. This cytokine is expressed and acts on many cells participating in allergic inflammation such as antigen-experienced Th2 cells, mast cells, eosinophils, basophils, dendritic cells, and CD34+ stem cells, and induces Th2-type cytokine production by different cells; thus, these facts make a base for the assumption that IL-33 may play an important role in the pathogenesis of allergic inflammation such as allergic rhinitis [24, 33].
Mast Cells
IL-33 alone or together with TSLP directly acts upon mature as well as precursor human mast cells [34]. IL-33 is able to induce the release of proinflammatory cytokines in bone marrow-derived mast cells [29, 35]. IL-33 activates mast cells as a result of its release from injured structural cells. Mast cells through ST2 receptor recognize IL-33, and signaling pathways are started with subsequent activation of NFκB and transcription of proinflammatory cytokines such as interleukins IL-1β, IL-6, IL-8, IL-13, TNF, chemokines, and prostaglandins [12•]. IL-33 may induce cytokine production in either the presence or absence of co-stimulation of mast cells via IgE/antigen–FεRI signals [35, 36]. IL-33 primes mast cells for activation by IgG immune complexes, and enhances its survival and adhesion [37].
Basophils
IL-33 also exerts distinct effects on basophils. IL-33/ST2 interactions enhanced degranulation in response to IgE cross-linking stimuli and enhanced basophil migration to eotaxin without effect on surface expression of CCR3 [22, 24]. Moreover, IL-33 synergizes with IL-3 to promote IL-4 production and CD11b expression by basophils, enhancing basophil adhesiveness [2•, 24]. Similarly to mast cells, human blood-derived basophils responded to IL-33 via ST2 receptor and as a result produce pro-inflammatory cytokines such as IL-1β, IL-4, IL-5, IL-6, IL-8, IL-13, and granulocyte macrophage colony-stimulating factor [24, 38, 39].
T Cells
IL-33 induces Th2 cytokine production and chemotaxis of in vitro polarized Th2 cells [5, 12•, 40]. In mouse airways, antigen-specific Th2 cells stimulated with IL-33 preferentially induce production of IL-5 and IL-13, but not IL-4, thus these T cells are called atypical Th2 cells [41•]. Similar results were obtained in BAL fluid after intra-nasal administration of IL-33 in which IL-5 and IL-13 levels increased and IL-4 did not change, suggesting that IL-33 may be involved in IL-4-independent Th2 cell differentiation [13]. IL-33 enhances IL-5 and IL-13 and, interestingly, IFN-gamma production by human Th2 in vitro skewed cells in HDM-specific T cell culture. IL-33 was found to be involved in the mechanism of Th2 response to house dust mite allergens in experimental model of asthma and to peanut allergens in experimental model of food allergy and anaphylaxis [42]. However, in ST2-deficient and IL-33-deficient mice, normal Th2 cells differentiation was observed [43, 44]. Moreover, iNKT cells express ST2L receptor and IL-33 stimulates the production of both type 1 and type 2 cytokines [24]. Thus, IL-33 promotes a Th2-type response and under certain conditions a Th1-type response as well.
Eosinophils
IL-33 mediates blood eosinophilia and eosinophil airway inflammation in mice [45]. It stimulates eosinophils differentiation from CD117+ progenitors in an IL-5-dependent manner. In humans, it enhances eosinophils survival, upregulates cell surface expression of adhesion molecule ICAM-1 on eosinophils, and suppresses ICAM-3 and selectin. IL-33 mediates significant release of the proinflammatory cytokine IL-6 and the chemokines CXCL8 and CCL2 and enhances Siglec-8-mediated apoptosis of eosinophils [46, 47]. In mice treated with IL-33, increased expression of CCR3 and CCR4 and their ligands, CCL11, CCL17, and CCL22, implicated in eosinophils chemotaxis in lung tissue was observed [13]. In knock-out IL-33 mice, eosinophils infiltration and cytokine production were reduced [13]. IL-33 stimulates eosinophils superoxide production and degranulation [48]. In humans, a correlation between blood and pulmonary eosinophilia with elevated IL-33 serum level was found [49].
IL-33 and Allergic Rhinitis
The plenipotent and pleiotropic activity of IL-33 in immune reactivity skewed into the Th2-type highlights the role of this protein in allergy. The pathogenesis of allergic rhinitis is driven by Th2-type immune response. Thus, the pertinence of IL-33 in this disorder can be safely assumed. There have been recently published papers which support the clinical relevance of this notion.
The results of our study concerning the issue showed elevated IL-33 serum level in patients with intermittent allergic rhinitis sensitive to tree and/or grass pollen. The increased level of this protein was comparable with that observed in asthma patients. Moreover, the elevated level of this protein was correlated with the severity of rhinitis symptoms, suggesting the role of this protein in the pathophysiology of this disorder. However, unexpectedly, ST2 serum level was in the same range as in healthy controls and in patients suffering from asthma [27]. It is notable the asthma patients included in the study were in the stable phase of the disease; the symptoms were well-controlled by low doses of inhaled corticosteroids and short and/or long acting b2-mimetics.
As presented above, IL-33 mediates its biological effects via interactions with a receptor consisting of two components, ST2 and IL-1 receptor accessory protein. The ST2/IL-33 pathway is associated with Th2-type pathology both in the promotion and maintenance of allergic inflammation. On the other hand, soluble ST2 being essential for the function of IL-33 acts as a decoy receptor for IL-33 bioactivities [50]. These data explain, at least to some extent, the discrepancy between ST2 and IL-33 serum level in our patients. Importantly, the results of other studies have demonstrated significant increases in the ST2 serum level in asthma exacerbation episodes both in adults and children [25, 26].
Regarding these observations, one can assume that soluble ST2-dependent regulation of IL-33 bioactivity occurs in intense airway inflammation. It is the most profound feature of symptomatic asthma but not rhinitis.
Our data remain in agreement with the observation of Sakashita et al. [51] indicating significant increased serum levels of IL-33 in patients suffering from intermittent allergic rhinitis caused by Japanese cedar pollen sensitization. The higher levels of IL-33 in nasal secretions from house dust mite-sensitive patients with perennial allergic rhinitis was observed [52]. Moreover, the genetic polymorphism in ST2 and IL-33 has been proved in rhinitis [5, 18]. These data further support the concept of the role of a ST2/IL-33 axis in the pathogenesis of this disorder.
The role of IL-33 in rhinitis was also pointed out in an excellent study published by Haenuki et al. [35]. They proved in an experimental study performed in a murine model that IL-33 constitutively expressed in nasal epithelial cells is released after allergen exposure. The diminished IL-33 expression after exposure to allergen was associated with the increase of this protein in nasal lavage fluid. This justifies the measuring of IL-33 levels in nasal lavage fluid after allergen provocation as a marker of this pathology. In allergen-immunized allergen-challenged IL-33−/− mice ,significant diminishment in sneezing and accumulation of eosinophils and basophils in the nasal mucosa, parallel with the reduction in specific IgE response, was proved. Furthermore, histological examinations revealed only slight, if any, changes in the IL-33−/− mice after allergen provocation. On the contrary, both early and late phase reactions relevant for rhinitis symptoms were markedly developed in allergen-immunized and then allergen-challenged IL-33+/+ mice. These results evidenced the contribution of IL-33 in the pathogenesis of rhinitis. It is important because the allergen-specific murine model mimics the main characteristics of human rhinitis. Significant upregulation of IL-33 mRNA expression in inflamed nasal epithelial cells of patients with intermittent allergic rhinitis during pollen season was observed, as in the allergic rhinitis murine model.
Prompt IL-33 release from the epithelial cells upon allergen exposure is essential for allergic rhinitis symptoms, through the bioactivity of the histamine released from the mast cells after cross-linkage of FceRI with allergen-specific IgE and infiltration of eosinophils/basophils and mastocytes in the nasal mucosa as in the site of allergen challenge. The ongoing inflammation process is directly responsible for the late phase reaction and the chronic symptoms of the disease. This also applies to allergic rhinitis. In turn, cytokines produced by IL-33- stimulated Th2 cells can induce goblet cells hyperplasia which might lead to the irreversible mucosal hypertrophy also seen in patients with allergic rhinitis [12•, 35].
IL-33 functioning as an “alarmin”, secreted following epithelial damage from different pathogens, plays a central role in the Th2 cytokine-mediated eosinophilic inflammation. As such, IL-33 is implicated in allergic respiratory pathology, not only for rhinitis but also for asthma [6, 7, 12•, 53].
Important data with plausible significant potential of this protein for human pathology have been presented by other authors. Anti-IL-33 antibody was found to be effective in suppression of symptoms of the mouse model ovalbumin-induced allergic rhinitis. The therapeutic effect of the anti–IL-33 antibody in experimental allergic rhinitis (non–scratching behavior) was confirmed by the decreased eosinophilic infiltration in the nasal mucosa, with a decrease in the number of eosinophils as well as the titers of IL-4, IL-5, and IL-13 cytokines in BAL fluid [54•]. Similar results were obtained in a soluble form of ST2-treated animals in such a model [55]. On the other hand, intra-nasal administration of IL-33 triggers allergic inflammation evolvement in the airways [13]. Liu et al. [56] showed, in the murine model of allergic asthma, the potential of a rabbit polyclonal antibody against IL-33 to inhibit airway inflammation. It is particularly important in the light of the recently proposed “united airway disease” concept.
These data further speak for the profound ability of anti–IL-33 to suppress allergic inflammation ongoing in target organs and to be responsible for allergy symptoms. The results presented allow a belief in the potential applicability of this type of treatment in human respiratory allergies.
Discussing the role of IL-33 in airway allergy, it is worth mentioning the results of the study in the animal model which showed the association of IL-33 polymorphism with asthma but, against expectations, not atopy [57]. These observations are consistent with findings of other authors depicting no difference in IL-33 serum level between atopic and nonatopic asthma [58]. It remains in line with the characteristic of the IL-33 bioactivity. This cytokine is able to induce and maintain IgE-dependent inflammatory reaction in the absence of a specific allergen. It markedly corresponds with the evidence of IL-33 playing a crucial role in the innate eosinophilic airway inflammation, but not in acquired immune responses such as IgE production [45]. An elegant experimental mice model of allergic rhinitis discussed above [35] provides further an important insight into the role of IL-33 in an IgE-mediated immune response and allergic symptoms development. The allergen challenge of allergen-immunized IL-33−/− mice causes a considerable increase of total IgE level, but not rhinitis symptoms. The IL-33−/− mice evidenced elevated serum IgE concentrations in the immunization phase, but a simultaneously reduced capacity to increase IgE response on allergen challenge.
Thus, these results indicate that IL-33 has substantial role in the allergic inflammation development in the nasal mucosa closely related to the symptoms of the disease. Simultaneously, they proved that the role of IL-33 in the IgE production is dispensable [59]. Recruitment of inflammatory cells, mast cells, eosinophils, and basophils as well as Th2 cells into nasal mucosa is critical for rhinitis symptoms, particularly for its chronicity. In this process, IL-33 plays a substantial role, acting as a potent Th2 chemoattractant and stimulator of FceRI cells to release chemoattractant substances for eosinophils/basophils.
Although a limited number of exogenous and endogenous agents are capable of inducing Th2 (eliciting pro-TH2 gene expression) by epithelial cells, many cases of asthma and/or rhinitis are in nonatopic conditions. Proving the role of IL-33/ST2 as a signal to the mast cells degranulation in the absence of specific allergen explains these observations, at least to some extent [60].
IL-33 seems to be an increasingly useful marker of Th2-mediated diseases such as allergic rhinitis and bronchial asthma, although not of a specific character. IL-33 is produced by epithelial cells when they become injured or necrotic [7]. So IL-33 could be viewed as a marker of tissue injury, particularly in epithelial and/or endothelial cells which are directly exposed to environmental stimuli [6, 16]. The question is whether an allergen is able to cause necrosis and/or injury of the epithelial cells? It might be the case with regard to allergens having enzymatic activity. Further studies are needed to conclusively answer this question.
Nevertheless, there are data showing that inhalation of papain or house dust mites can cause airway inflammation without prior sensitization. This phenomenon is associated with increased IL-33 mRNA expression in the absence of acquired immune cells. The same is observed after administration of recombinant IL-33 [12•, 61].
Conclusions
Observations regarding the role of IL-33 in disturbed immune regulation provide novel insights into the mechanism of allergic diseases. IL-33 is released by many cells during tissue injury upon proinflammatory stimulation, promoting the severity of allergies.
The increase level/activity of IL–33 in allergic disorders and its correlation with disease severity on the one hand and, on the other hand, the effectiveness both of an anti-IL-33 antibody and a soluble form of ST2 in alleviating signs and symptoms of ovalbumin-induced allergic rhinitis/asthma in an animal model justify the view of IL-33 as a potential therapeutic target in allergic disease.
It is important that the antihistamines commonly applied in the treatment of allergic rhinitis are potent to down-regulate the expression of IL-5 mRNA and histamine H1-receptor gene expression, but not that of IL-33 mRNA in the mucosa of the respiratory tract [62]. There is a similar case with regard to the corticosteroids, the drugs of first choice both in the treatment of allergic rhinitis and asthma. Corticosteroids exert their activity only partially on IL-33-mediated immune reaction [63].
One can assume that the down-regulation of IL-33 gene expression in the nasal mucosa provides the basis for a better therapeutic effect of allergic rhinitis treated with antihistamines and/or corticosteroids.
This therapeutic concept is particularly attractive in the light of the observations of the baseline IL-33 mRNA expression in epithelial cells derived from chronic rhinosinusitis with nasal polyps, which was several times higher in recalcitrant patients than in those with responsive disease [64•]. In the light of these data, anti-IL-33 therapy seems to be particularly promising. These findings can be extrapolated to rhinitis and/or asthma, especially to those forms which are not sufficiently responsive to recently applied treatment, although there is a need for further conclusive research.
IL-33 is a potent proinflammatory cytokine activated in allergen- and non-allergen-dependent fashion. Its role is more potent in the elicitation phase of allergic reaction than in the sensitization phase. One might believe that the IL-33/ST2 pathway is particularly important for inflammation development in the late phase response. The results of the study performed in allergen-immunized mice with the use of recombinant human ST2-Fc (receptor of IL-33) chimera protein are encouraging [65].
Thus, considering the IL-33/ST2 pathway as a therapeutic target should be of particular importance. In this context, it is reasonable to believe in IL-33 as a novel therapeutic target for allergic disorders, not only in allergic rhinitis but also or even especially in asthma, which remains treatable but not curable until now due to a sustained inflammation process in the airway.
References
Recently published papers of particular interest have been highlighted as: • Of importance
Miller A. Role of IL-33 in inflammation and disease. J Inflammation. 2011;8:22.
• Lloyd CM. IL-33 family members and asthma – bridging innate and adaptive immune responses. Curr Opin Immunol. 2010;22:800–806. A comprehensive review on the role of IL-33 in immune response in asthma.
Onda H, Kasuya H, Takakura K, Hori T, Imaizumi T, Takeuchi T, Inoue I, Takeda J. Identification of genes differentially expressed in canine vasospastic cerebral arteries after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 1999;19:1279–88.
Baekkevold ES, Roussigné M, Yamanaka T, Johansen FE, Jahnsen FL, Amalric F, Brandtzaeg P, Erard M, Haraldsen G, Girard JP. Molecular characterization of NF-HEV, a nuclaer factor preferentially expressed in human high endothelial venules. Am J Pathol. 2003;163:69–79.
Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an interleukin 1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90.
Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel “alarmin”? PLoS One. 2008;3:e3331.
Lamkanfi M, Dixit VM. IL-33 raises alarm. Immunity. 2009;31:5–7.
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98.
Cayrol C, Girard JP. The IL-1-like cytokine IL-33 is inactivated after maturation of caspase-1. Proc Natl Acad Sci USA. 2009;106:9021–6.
Talabot-Ayer D, Lamacchia C, Gabay C, Palmer G. Interleukin-33 is biologically active independently of caspase-1 cleavage. J Biol Chem. 2009;284:19420–6.
• Ohno T, Morita H, Arae K, Matsumoto K, Nakae S. Interleukin-33 in allergy. Allergy 2012;67:1203–1214. This study presents a comprehensive up-to-date analysis regarding the role of IL-33/ST2 axis in the pathogenesis of allergy.
Louten J, Rankin A, Li Y, Murphy E, Beaumont M, Moon C, Bourne P, McClanahan TK, Pflanz S, de Waal Malefyt R. Endogenous IL-33 enhances Th2 cytokine production and T-cell response during allergic airway inflammation. Int Immunol 2011;23307–315
Haraldsen G, Balogh J, Pollheimer J, Sponheim J, Kuchler AM. Interleukin-33cytokine of dual function or novel alarmin? Trends Immunol. 2009;30:227–33.
Carriere V, Roussel L, Ortega N, Lacorre DA, Amirich L, Aguilar L, et al. IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci USA. 2007;104:282–7.
Tago K, Noda T, Hayakawa M, Iwahana H, Yanagisawa K, Yashiro T, Tominaga S. Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2 V. Biochem Biophys Res Commun. 2001;285:1377–83.
Hong J, Bae S, Jhun H, Lee S, Choi J, Kang T, Kwak A, Hong K, Kim E, Jo S, Kim S. Identification of constitutively active interleukin 33 (IL-33) splice variant. J Biol Chem. 2011;286:20078–86.
Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy. 2002;32:1520–6.
Coyle AJ, Lloyd C, Tian J, Nguyen T, Erikkson C, Wang L, Ottoson P, Persson P, Delaney T, Lehar S, et al. Crucial role of the interleukin 1 receptor family member T1/ST2 in T helper cell type 2-mediated lung mucosal immune responses. J Exp Med. 1999;190:895–902.
Xu D, Chan WL, Leung BP, Huang FP, Wheeler R, Piedrafita D, Robinson JH, LIew FY. Selective expression of a stable cell surface molecule on type 2 but not type 1 helper T cells. J Exp Med. 1998;187:787–94.
Lohning M, Stroehmann A, Coyle AJ, et al. T1/ST2 is preferentially expressed on murine Th2 cells, independent of interleukin 4, interleukin 5, and interleukin 10, and important for Th2 effector function. Proc Natl Acad Sci USA. 1998;965:6930–5.
Chan WL, Pejnovic N, Lee CA, Al-Ali NA. Human IL-18 receptor and ST2L are stable and selective markers for the respective type 1 and type 2 circulating lymphocytes. J Immunol. 2001;167:1238–44.
Joshi AD, Oak SR, Hartigan AJ, Finn WG, Kunkel SL, Duffy KE, Das A, Hogaboam CM. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. BMC Immunol. 2010;11:52.
Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–30.
Oshikawa K, Kuroiwa K, Tago K, Iwahana H, Yanagisawa K, Ohno S, Tominaga SI, Sigiyama Y. Elevated soluble ST2 protein levels in sera of patients with asthma with an acute exacerbation. Am J Respir Crit Care Med. 2001;164:277–81.
Ali M, Zhang G, Thomas WR, et al. Investigations into the role of ST2 in acute asthma in children. Tissue Antigens. 2009;73:206–12.
Glück J, Rymarczyk B, Rogala B. Serum IL-33 but not ST2 level is elevated in intermittent allergic rhinitis and is a marker of the disease severity. Inflamm Res. 2012;5:547–50.
Kamekura R, Kojima T, Takano K, Go M, Sawada N, Himi T. The role of IL-33 and its receptor ST2 in human nasal epithelium with allergic rhinitis. Clin Exp Allergy. 2012;42:218–28.
Ali S, Huber M, Kollewe C, BIschoff SC, Falk W, Martin MU. IL-1 receptor accessory protein is Essentials for IL-33-induced activation of T lymphocytes and mast cells. Proc Natl Acad Sci USA. 2007;104:18660–5.
Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, Girard JP. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel IL-33-LacZ gene trap reporter strain. J Immunol. 2012;188:3488–95.
Zhang L, Lu R, Zhao G, Pflugfelder SC, Li DQ. TLR-mediated induction of pro-allergic cytokine IL-33 in ocular mucosal epithelium. Int J Biochem Cell Biol. 2011;43:1383–91.
Wisniewski JA, Borish L. Novel cytokines and cytokine-producing T cells in allergic disorders. Allergy Asthma Proc. 2011;32:83–94.
Oliphant C, Barlow JL, McKenzie A. Insights into the initiation of type 2 immune responses. Immunology. 2011;134:378–85.
Allakhverdi Z, Smith DE, Comeau M, Delespesse G. Cutting Edge: The ST2 ligand Il-33 potently activates and drives maturation of human mast cells. J Immunol. 2007;179:2051–4.
Haenuki Y, Matsushita K, Futatsugi-Yumikura S, Ishii KJ, Tatsukata K, Yoshimasa I, Shigeharu F, Yasuda M, et al. A critical role of IL-33 in experimental allergic rhinitis. J Allergy Clin Immunol. 2012;130:184–94.
Iikura M, Suto H, Kajiwara N, Oboki K, Ohno T, Okayama Y, Saito H, Galli SJ, Nakae S. IL-33 can promote survival, adhesion and cytokine production in human mast cells. Lab Invest. 2007;87:971–8.
Kaieda S, Wang JX, Shnayder R, Fishgal N, Nei H, Lee RT, Stevens RL, Nigrovic PA. Interleukin-33 primes mast cells for activation by IgG immune complexes. PLoS One. 2012;7:e47252.
Suzukawa M, Likura M, Oketsu R, Nagase H, Tamura C, Komiya A, Nakae S, Matsuhima K, Ohta K, Yamamto K, Yamaguchi M. An IL-1 cytokine member, IL-33, induces human basophil activation via its ST2 receptor. J Immunol. 2008;181:5981–9.
Pekaric-Petkovic T, Didichenko SA, Kaempfer S, Spiegl N, Dahinden CA. Human basophils and eosinophils are the direct targer leukocytes of the novel IL-1 family member IL-33. Blood. 2009;113:5981–9.
Komai-Koma M, Xu D, Li Y, McKenzie AN, McInnes IB, Liew FY. IL-33 is a chemoattractant for human Th2 cells. Eur J Immunol. 2007;37:2779–86.
• Kurowska-Stolarska M, Kewin P, Murphy G et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol 2008;181:4780–4790. In this study it was show that IL-33 may be involved in IL-4 independent Th2 cell differentiation.
Chu DK, Llop-Guevara A, Walker TD, Flader K, Goncharova S, Boudreau JE, Moore CL, Waserman S, Coyle AJ, Kolbeck R, Humbles AA, Jordana M. IL-33, but not thymic stromal lymphopoietin or IL-25, is central to mite and peanut allergic sensitization. J Allergy Clin Immunol. 2013;131:187–200.e8.
Kashiwamura S, Kuribayashi K, et al. The absence of interleukin 1 receptor-related T1/ST2 does not affect T helper cell type 2 development and its effector function. J Exp Med. 1999;190:1541.
Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069–76.
Stolarski B, Kurowska-Stolarska M, Kewin P, Xu D, Liew FY. IL-33 exacerbates eosinophils-mediated airway inflammation. J Immunol. 2010;185:3472–80.
Chow JY, Wong CK, Cheung PF, Lam CW. Intracellular signaling mechanisms regulating the activation of human eosinophils by the novel Th2 cytokine IL-33: implications for allergic inflammation. Cell Mol Immunol. 2010;7:26–34.
Na HJ, Hudson SA, Bochner BS. IL-33 enhances Siglec-8 mediated apoptosis of human eosinophils. Cytokine. 2012;57:169–74.
Cherry WB, Yoon J, Bartemes KR, Lijima K, Kita H. A novel IL-1 family cytokine, IL-33, potently activates human eosinophils. J Allergy Clin Immunol. 2008;121:1484–90.
Kim H, Jun C, Lee Y, et al. Levels of circulating IL-33 and eosinophil cationic protein in patients with hypereosinophilia or pulmonary eosinophilia. J Allergy Clin Immunol. 2010;126:880.
Oboki K, Ohno T, Kajiwara N, Saito H, Nakae S. Il-33 and Il-33 receptors in host defense and diseases. Allergol Int. 2010;59:149–60.
Sakashita M, Yoshimoto T, Hirota T, Harada M, Okubo K, Osawa Y, et al. Association of serum interleukin-22 level and the interleukin-33 genetic variant with Japanese Cedar pollinosis. Clin Exp Allergy. 2008;38:1875–81.
Asaka D, Yoshikawa M, Nakayama T, Yoshimura T, Moriyama H, Otori N. Elevated Levels of Interleukin-33 in the Nasal Secretions of Patients with Allergic Rhinitis. Int Arch Allergy Immunol. 2012;158:47–50.
Eiwegger T, Akdis CA. IL-33 links tissue cells, dendritic cells and Th2 cell development in a mouse model of asthma. Eur J Immunol. 2011;41:1535–8.
• Kim Y.H, Yang T.Y, Park C.S, Ahn S.H, Son B.K, Kim J.H, Lim D.H, Jang T.Y. Anti-IL-33 antibody has a therapeutic effect in a murine model of allergic rhinitis. Allergy 2012;67:183–190. The results of this experimental study provide an excellent framework for further research aimed at the improvement of allergy inflammation treatment.
Kearley J, Buckland KF, Mathic SA, Lloyd CM. Resolution of allergic inflammation and airway hyperreactivity is dependent upon disruption of the T1/ST1-IL-33 pathway. Am J Respir Crit Care Med. 2009;179:772–81.
Liu X, Li M, Wu Y, Zhou Y, Zeng L, Huang T. Anti-IL-33 antibody treatment inhibitis airway inflammation in a murine model of allergic asthma. Biochem Biophys Res Commun. 2009;386:181–5.
Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med. 2010;363:1211–21.
Komai-Koma M, Brombacher F, Pushparaj PN, Arendse B, McSharry C, Alexander J, Chaudhuri R, Thomson NC, McKenzie ANJ, McInnes I, Liew FY, Xu D. Interleukin-33 amplifies IgE synthesis and triggers mast cell degranulation via interleukin-4 in naïve mice. Allergy. 2012;67:1118–26.
Ho LH, Ohno T, Oboki K, Kajiwara N, Suto H, Iikura M, et al. IL-33 induces IL-13 production by mouse mast cells independently of IgE-FcepsilonRI signals. J Leukoc Biol. 2007;82:1481–90.
Kondo Y, Yoshimoto T, Yasuda K, Futatsugi-Yumikura S, Morimoto M, Hayashi N, et al. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lungs in the absence of adaptive immune system. Int Immunol. 2008;20:791–800.
Pushparaj PN, Tay HK, H’ng SC, Pitman N, Xu D, McKenzie A, et al. The cytokine interleukin-33 mediates anaphylactic shock. Proc Natl Acad Sci USA. 2009;106:9773–8.
Kitamura Y, Mizuguchi H, Ogishi H, Kuroda W, Hatttori M, Fukui H, Takeda N. Preseasonal prophylactic treatment with antihistamines suppresses Il-5 but not Il-33 mRNA expression in the nasal mucosa of patients with seasonal allergic rhinitis caused by Japanese cedar pollen. Acta Oto-Laryngologica. 2012;132:434–8.
Préfontaine D, Lajoie-Kadoch S, Foley S, Audusseau S, Olivenstein R, Halayko AJ, Lemière C, Martin JG. Hamid Q1 Increased expression in severe asthma: evidence of expression by airway smooth muscle cells. J Immunol. 2009;15:5094–103.
• Reh D, Wang Y, Ramanathan M, Lane AP. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy 2010;24:105–109. The first study to show evidence that Il-33 is produced by sinonasal epithelial cells derived from chronic sinusitis with nasal polyps, and Il-33 mRNA expression is several higher in patients with recalcitrant disease than those with a responsive disorder.
Leung BP, Xu D, Culshaw S, Mcinnes IB, Liew FY. A novel therapy of murine collagen – induced arthritis with soluble T1/ST2. J Immunol. 2004;173:145–50.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Rogala, B., Glück, J. The Role of Interleukin-33 in Rhinitis. Curr Allergy Asthma Rep 13, 196–202 (2013). https://doi.org/10.1007/s11882-013-0338-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11882-013-0338-z