
Abstract
A practical and step-economic route to Favipiravir, an antiviral drug, was developed. Favipiravir was synthesized in only six steps from 3-aminopyrazine-2-carboxylic acid with an overall yield of about 22.3%. Key intermediates 3 and 6 were obtained in excellent purity via recrystallization from optimized solvents, which was beneficial to large-scale production. In the key synthetic reaction, 3,6-dichloropyrazine-2-carbonitrile (6) was reacted sequentially, in one pot, with KF and 30% H2O2 to give (after crystallization from 95% EtOH) favipiravir as colorless crystals, with a 60% yield for this final step of the synthesis.
Similar content being viewed by others


Introduction
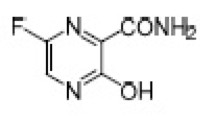
Favipiravir (T-705; 6-fluoro-3-hydroxy-2-pyrazinecarboxamide), a RNA-dependent RNA polymerase selective inhibitor, is a novel broad-spectrum antiviral drug (Furuta and Egawa 2000; Zhao et al. 2015). A range of RNA virus such as West Nile virus (Morrey et al. 2008), Norwalk virus (Rocha-Pereira et al. 2012), and Ebola virus (Oestereich et al. 2014) could be inhibited by T-705. Presently, it was also reported that postexposure administration of T-705 was effective against encephalitis viruses (Yamada et al. 2016). Accordingly, substantial efforts have been made toward the development effective and practical methods to synthesize T-705 (Zhang et al. 2017).
T-705 was first synthesized by Japanese Toyama Chemical Co., Ltd. in 2000. However, when Beldar et al. (2009) prepared T-705 according to this previous procedure, they encountered problems that required optimization of the original synthetic procedures. Afterward, additional modifications to the synthetic route were reported (Hara et al. 2011; Caldwell et al. 2012; Zhang et al. 2013; Shi et al. 2014; Furuta and Egawa 2000; Zheng et al. 2012a, b; Wang et al. 2014, 2015; Bao et al. 2012). In 2013, T-705 was successfully prepared by Zhang’s group from 3-amino-2-pyrazinecarboxylic acid with a good overall yield of about 22% in eight steps. Wang’s group (2014) reported another practical route to T-705 starting from amino malonic acid diethyl ester hydrochloride, but with poor overall yield of about only 9%. Very recently, Wang et al. (2015) developed a new synthetic route to T-705. However, the step of nitration and low overall yield (5.6%) imposed restrictions on its large-scale production. Despite these limitations, the synthesis of T-705 was carried out, although hampered by limited availability of the starting materials, long steps, low yield, and chromatography to purify the intermediates. Therefore, the development of a practical and step-economic route to Favipiravir with good yield is of great interest.
Among the reported synthetic routes to T-705, the Zhang’s method was particularly interesting due to its mild reaction conditions and good yield (Scheme 1). Nevertheless, there were eight steps in this synthetic procedure in which chromatography was needed to purify the crude products of the steps of fluorination, chlorination and hydrolysis. Driven by our continued interest in practical large-scale manufacture with low cost and high yield, we report here a modified procedure (Scheme 1). First, we observed that the bromination step introduced impurities and made it very difficult to purify the products of the following steps, leading to the need for chromatography for post-processing. Consequently, the strategy of recrystallization was applied to replace the chromatography. The conditions for recrystallization were optimized here and pure brominated product was obtained successfully. Meanwhile, the method of recrystallization was also applied to purify compound 6. Therefore, in our modified route, all purification was by recrystallization. In addition, to reduce the cost and improve the overall yield, the synthetic steps were shorted to six steps using a one-pot strategy of the last three steps. The overall yield of this step was increased by about 20%.

Synthetic route of T-705 reported by Zhang and modified by us
Experimental
Materials and methods
Unless otherwise noted, all the reagents were purchased from commercial suppliers and used without further purification. All reagents were weighed and handled in the air at room temperature. All reactions were monitored by thin-layer chromatography (TLC) on 25.4 × 76.2 mm silica gel plates (GF-254). NMR spectra were recorded on Brucker AVANCE III 400/500 NMR spectrometer. Chemical shifts were reported in parts per million (ppm, δ) relative to tetramethylsilane. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Mass spectra were recorded on Thermo Finnigan-MAT 95 XP instrument. Melting points (m.p.) were measured by Büchi 510 melting point apparatus and are uncorrected.
Preparation of methyl 3-aminopyrazine-2-carboxylate (2) (Caldwell et al. 2012)
To a suspension of 3-aminopyrazine-2-carboxylic acid (20 g, 1 eq) in methanol (200 mL) in an ice bath, concentrated H2SO4 (20 mL) was added slowly (dropwise, over at least a 2 h interval). After vigorous stirring for 48 h, a black solution was formed and TLC (EA:PE = 1:2, v/v, R f = 0.22) analysis showed the complete consumption of compound 1. The reaction mixture was then concentrated. The mixture was adjusted to pH 7–8 by progressively adding Na2CO3 (20%, m/m) aqueous solution (100 mL) under ice conditions. The mixture was filtered. The filter residue was collected and dried under reduced pressure to obtain black solid 2 (15.4 g, 70%).
Methyl 3-aminopyrazine-2-carboxylate (2)
Yield: 70%, black solid. m.p.: 173–175 °C (Lit. 172–173 °C, Caldwell et al. 2012). 1H-NMR (400 MHz, DMSO-d6): δ 8.26 (d, J = 2.2 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.37–7.32 (m, 2H), 3.84 (s, 3H). 13C-NMR (101 MHz, DMSO-d6): δ 166.37, 155.78, 147.72, 132.40, 123.12, 52.00. MS (EI): m/z = 153.1 (M+). 94.1 (M+, –COOCH3).
Preparation of methyl 3-amino-6-bromopyrazine-2-carboxylate (3)
Methyl 3-aminopyrazine-2-carboxylate (15 g, 1 eq) was suspended in 150 mL acetonitrile and NBS (18.3 g, 1 eq) was added in batches under the protection of N2. Then, the solution was stirred at room temperature for 24 h. Subsequently, the mixture was adjusted to pH 7–8 which was monitored by extensive pH indicator paper by progressively adding Na2CO3 (20%, m/m, 10 mL) aqueous solution at 0 °C. Then, the solution was filtered. The filter residue was collected and dried under reduced pressure to obtain a brown-colored solid. The crude product was then dissolved into 0.8 L dichloromethane by heating to reflux for 30 min. The suspension was filtered to remove the insoluble impurities. Then, the solvent was removed under reduced pressure. Afterward, the residue was redissolved into 0.7 L EtOH (95%). The suspension was heated to reflux for 30 min and filtered to crystallize. Then, the filtrate was cooled to 25–30 °C which was kept for 4 h. The crystal of pure 3 (19.7 g, 87%) (TLC, EA:PE = 1:2, v/v, R f = 0.64) could be obtained in open system.
Methyl 3-amino-6-bromopyrazine-2-carboxylate (3)
Yield: 87%, yellow crystals, granular, m.p.: 174–176 °C (Lit. 175.3–175.9 °C) (Caldwell et al. 2012). 1H-NMR (400 MHz, DMSO-d6): δ 8.43 (s, 1H), 7.57 (s, 2H) 3.86 (s, 3H). 13C-NMR (101 MHz, DMSO-d6): δ 165.27, 154.86, 150.04, 122.56, 122.31, 52.24. MS (EI): m/z = 232.0 (M+, Br79, 45), 234.0 (M+, Br81, 45), 201.0 (M+, –OCH3, Br79, 30), 203.0 (M+, –OCH3, Br81, 20), 173.0 (M+, –COOCH3, Br79, 100), 175.0 (M+, –COOCH3, Br81, 100), 146.0 (Br79, 15), 148.0 (Br81, 10).
Preparation of methyl 3-hydroxy-6-bromopyrazine-2-carboxylate (4) (Furuta and Egawa 2000)
Methyl 3-amino-6-bromopyrazine-2-carboxylate (10 g, 1 eq) was suspended in concentrated H2SO4 (40 mL), and then, NaNO2 (5.96 g, 2 eq) was added in batches at −5 to 0 °C, after which the reaction was stirred at 25 °C for 2 h until the solid was all dissolved. Then, the reaction solution was poured into ice water (400 mL) and vigorously stirred for 1.5 h. Afterward, the solution was extracted with ethyl acetate (3 × 50 mL). The organic layer was combined and washed with water (3 × 30 mL). Then, it was dried with anhydrous Na2SO4 for 5 h and filtered to remove the drying agent. The ethyl acetate was removed by distillation under reduced pressure to obtain methyl 3-hydroxy-6-bromopyrazine-2-carboxylate (9.3 g, 92.7%) (TLC, EA:MeOH = 1:1, v/v, R f = 0.77).
Methyl 3-hydroxy-6-bromopyrazine-2-carboxylate (4)
Yield: 92.7%, gray solid, m.p.: 120–122 °C (Lit. 120.5–121.5 °C) (Caldwell et al. 2012). 1H-NMR (400 MHz, DMSO-d6): δ 8.40 (s, 1H), 3.85 (s, 3H). 13C-NMR (101 MHz, DMSO-d6): δ 163.20, 157.11, 143.89, 134.81, 122.06, 52.43. MS (EI): m/z = 233.0 (M+, Br79, 40), 235.0 (M+, Br81, 40), 202.0 (M+, –OCH3, Br79, 45), 204.0 (M+, –OCH3, Br81, 25), 174.0 (M+, –COOCH3, Br79, 100), 176.0 (M+, –COOCH3, Br79, 60).
Preparation of 3-hydroxy-6-bromopyrazine-2-carboxamide (5) (Furuta and Egawa 2000)
Aqueous ammonia (50 mL) was added into methyl 3-hydroxy-6-bromopyrazine-2-carboxylate (5 g, 1 eq) and stirred at room temperature for 3 h. The solution was filtered and the filter residue was washed with 20 mL aqueous ammonia and pure 3-hydroxy-6-bromopyrazine-2-carboxamide (4.4 g, 94%) (TLC, EA:MeOH = 1:1, v/v, R f = 0.25) was obtained.
3-Hydroxy-6-bromopyrazine-2-carboxamide (5)
Yield: 94%, yellow solid, m.p.: 153–155 °C.1H-NMR (400 MHz, DMSO-d6): δ 10.29 (s, 1H), 7.98 (s, 1H), 7.40 (s, 2H). 13C-NMR (101 MHz, DMSO-d6): δ 166.82, 166.74, 147.70, 133.01, 117.99. MS (EI): m/z = 218.0 (M+, Br79, 95), 220.0 (M+, Br81, 5), 200.0 (M+, –H2O, Br79, 5), 202.0 (M+, –H2O, Br81, 5), 174.0 (M+, –CONH2, Br79, 35), 176.0 (M+, –CONH2, Br81, 20), 147.0 (Br79, 20), 149.0 (Br81, 18).
Preparation of 3, 6-dichloropyrazine-2-carbonitrile (6)
POCl3 (5.6 g, 4 eq) was added into 3-hydroxy-6-bromopyrazine-2-carboxamide (2 g, 1 eq). The mixture was heated to 70 °C for 15 min, resulting in a homogenous solution. The solution was cooled to ambient temperature and DIEA (3.57 g, 3 eq) was added dropwise, keeping the temperature below 60 °C. The reaction mixture was stirred at 60 °C (Zheng et al. 2012a, b) for 1 h, then at 80 °C for 1 h and finally stirred at 100 °C for at least 4 h. The cooled reaction mixture was poured into crush ice (500 g) with vigorously stirring for at least 1 h using TLC (EA:PE = 1:5, v/v, R f = 0.74) to monitor the reaction. The solution was extracted with ethyl acetate (5 × 30 mL), and the organic phase was washed with brine (3 × 20 mL), dried with anhydrous Na2SO4, and concentrated to dryness to obtain the crude 6. The crude compound of 6 was suspended into petroleum ether (b.p. = 60–90 °C, 60 mL). The suspension was heated to reflux for 30 min and was filtered. The filtrate was cooled to 25–30 °C which was kept for 4 h. Then, 3, 6-dichloropyrazine-2-carbonitrile (1.6 g, 70%) was obtained in open system.
3, 6-Dichloropyrazine-2-carbonitrile (6)
Yield: 70%, yellow crystals, rods, m.p.: 87–88 °C (Lit. 89.7–89.8 °C) (Caldwell et al. 2012). 1H-NMR (400 MHz, DMSO-d6): δ 9.04 (s, H). 13C-NMR (101 MHz, DMSO-d6): δ 149.05, 148.11, 146.40, 128.20, 113.51. MS (EI): m/z = 173.0 (M+, Cl35, Cl35, 100), 175.0 (M+, Cl35, Cl37, 75), 177.0 (M+, Cl37, Cl37, 10), 147.0 (M+, –CN, 10), 121.0 (M+, –CN, –2CH, 15).
Preparation of 6-fluoro-3-hydroxypyrazine-2-carboxamide (7)
KF·2H2O (1.3 g, 6 eq) and TBAB (tetrabutylammonium bromide) (0.3 g, 0.4 eq) were added into the mixed solvent containing 4 mL of DMSO and 8 mL of toluene. The toluene was subsequently removed by distillation under reduced pressure. A further 8 mL of toluene was then added and once again removed by distillation under reduced pressure with the purpose of removing the moisture of the reagent relating to the reaction. Then, 3, 6-dichloropyrazine-2-carbonitrile (0.4 g, 1 eq) was added and the mixture was stirred at 50 °C for 3 h. Subsequently, anhydrous K2CO3 (0.04 g) and 30% H2O2 (0.28 mL) were added into the solution at 0 °C stirring for 1.5 h at 25 °C. Water (1 mL) and NaHCO3 (0.132 g) were added into the 3,6-difluoropyrazine-2-carboxamide with magnetic stirring for 8 h at 50 °C and TLC (MeOH:EA = 1:1, v/v, R f = 0.72) was used to monitor the reaction. Then, 6 M HCl was added into the solution and the pH of the solution was adjusted to 1.0. Then, the solution was extracted with ethyl acetate (5 × 10 mL). The organic phase was washed with brine (3 × 10 mL), dried with anhydrous Na2SO4, and was filtered to remove the drying agent. The ethyl acetate was removed by distillation under reduced pressure to obtain the crude compound of 7. Then, the crude 7 was dissolved into EtOH (6 mL, 95%). The suspension was heated to reflux for 30 min and was filtered to crystallize. The filtrate was cooled to 25–30 °C which was kept for 4 h. The crystal of pure 6-fluoro-3-hyroxypyrazine-2-carboxamide (7) (0.25 g, 60%) was obtained in open system.
6-Fluoro-3-hydroxypyrazine-2-carboxamide (7)
Yield: 60%, colorless crystals, needles, m.p.: 175–177 °C, (Lit. 178.9–180.1 °C) (Caldwell et al. 2012). 19F-NMR (376 MHz, CDCl3): δ -92.79 (s, 1F). 1H-NMR (500 MHz, DMSO-d6): δ 13.41 (s, OH), 8.75 (s, 1H), 8.51 (d, J = 7.7 Hz, 2H). 13C-NMR (126 MHz, DMSO-d6): δ 168.66, 159.66, 152.32 (d, J = 243.4 Hz), 135.87, 122.09. MS (ESI): m/z = 158.1 [M+H]+, 180.0 [M+Na]+.
Results and discussion
The modified synthetic route was illustrated in Scheme 2. First, 3-aminopyrazine-2-carboxylic acid was chosen to be the starting material. Compound 3 was easily synthesized according to the reported literature (Zhang et al. 2013). To make this synthetic route more suitable for large-scale industry production, recrystallization was applied to purify the key intermediate methyl 3-amino-6-bromopyrazine-2-carboxylate (3). First, the solvents for recrystallization were screened (Table 1, entries 1–8). Pure 3 could not be recrystallized from many solvents, including CH2Cl2, EtOAc, MeOH, and EtOH (Table 1, entries 1–4). Fortunately, it was observed that compound 3 was freely dissolved into CH2Cl2 and the impurities were not. Therefore, compound 3 was obtained according to the preparation of methyl 3-amino-6-bromopyrazine-2-carboxylate (Table 1, entry 5). Sequentially, the volume of the solvent was considered (Table 1, entries 5–8). It was obvious that the quality of pure 3 increased with the increasing of the volume of solvent. The solvent of EtOH (95%) was green solvent and cheap to large-scale production. With pure 3 in hand, compound 6 was prepared rapidly. After optimizing solvents, the pure compound was also obtained by recrystallizing from petroleum ether.

Synthesis of 6-fluoro-3-hydroxypyrazine-2-carboxamide in this paper
Considering the compatibility of solvent in the last three steps, we reasoned that it was possible to simplify these steps by one-pot routing (Table 2, entries 1–3). First, according to Zhang’s synthetic route, the procedure of hydrolysis of nitrile was lastly carried out in the presence of H2SO4 (Table 2, entry 1). It was a mild catalyst-H2O2 that was used to complete the hydrolysis of nitrile in the previous literature (Katritzky et al. 1989) (Table 2, entry 2). Compound 7 could be obtained, but was difficult to separate and purify. Inspired by another reported literature (Beldar and Jordis 2009), we changed the sequence of the procedures of hydrolysis, fluorination, and nitrile aminolysis. The overall reaction went smoothly to give pure 7 with a good yield of 60% (Table 2, entry 3).
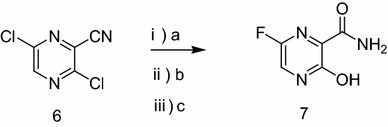
Conclusion
In summary, we demonstrated a facile route to synthesize 6-fluoro-3- hydroxypyrazine-2-carboxamide starting from commercial 3-aminopyrazine-2-carboxylic acid (1). A practical chromatography-free method of post-processing to refine 3-amino-6-bromopyrazine-2-carboxylate (3) and 3, 6-difluoropyrazine-2-carbonitrile (6) was offered, which is suitable for large-scale manufacture. The last three procedures were successfully carried out in one-step with high yield. This modified synthetic route was more efficient and economical with an overall yield of 22.3%.
References
Bao J, Huang H, Jiang Y, Zhang X (2012) A synthetic method of favipiravir. State Intellectual Property Office of the P.R.C. CN104496917A (issued April 8, 2012)
Beldar SV, Jordis U (2009) Synthetic studies towards the antiviral pyrazine derivative T-705. Institute of Applied Synthetic Chemistry, Vienna University of Technology, Vienna, p 13
Caldwell JJ, Veillard N, Collins I (2012) Design and synthesis of 2(1H)-pyrazinones as inhibitors of protein kinases. Tetrahedron 68:9713–9728. doi:10.1016/j.tet.2012.09.039
Furuta Y, Egawa H (2000) Nitrogenous heterocyclic carboxamide derivatives or salts thereof and antiviral agents containing both. European Patent Office WO, 00/10569 (issued March 2, 2000)
Hara T, Norimatsu N, Kurushima H, Kano T (2011) Method for producing dichloropyrazine derivative. United States Patent Application Publication US20110275817A1 (issued November 10, 2011)
Katritzky AR, Pilarski B, Urogdi L (1989) Cheminform abstract: efficient conversion of nitriles (I) to amides (II) with basic hydrogen peroxide in dimethyl sulfoxide. Synthesis 12:949–950. doi:10.1055/S-1989-27441
Morrey JD, Taro BS, Siddharthan V, Wang H, Smee DF, Christensen AJ, Furuta Y (2008) Efficacy of orally administered T-705 pyrazine analog on lethal West Nile virus infection in rodents. Antiviral Res 80:377–379. doi:10.1016/j.antiviral.2008.07.009
Oestereich L, Lüdtke A, Wurr S, Rieger T, Muñoz-Fontela C, Günther S (2014) Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res 105:17–21. doi:10.1016/j.antiviral.2014.02.014
Rocha-Pereira J, Jochmans D, Dallmeier K, Leyssen P, Nascimento MSJ, Neyts J (2012) Favipiravir (T-705) inhibits in vitro norovirus replication. Biochem Biophys Res Commun 424:777–780. doi:10.1016/j.bbrc.2012.07.034
Shi F, Li Z, Kong L, Xie Y, Zhang T, Xu W (2014) Synthesis and crystal structure of 6-fluoro-3-hydroxypyrazine-2-carboxamide. Drug Discov Ther 8(3):117–120. doi:10.5582/ddt.2014.01028
Wang H, Li X, Zhong W (2014) Synthesis of favipiravir. Chin J Pharm 45(11):1009–1012
Wang W, Liu M, Xiao X, Dai Q (2015) Synthesis of favipiravir. J Int Pharm Res 42(2):220–224. doi:10.13220/j.cnki.jipr.2015.02.018
Yamada K, Noguchi K, Komeno T, Furuta Y, Nishizono A (2016) Efficacy of favipiravir (T-705) in rabies postexposure prophylaxis. J Infect Dis 213(8):1253–1261. doi:10.1093/infdis/jiv586
Zhang T, Kong L, Li Z, Yuan H, Xu W (2013) Synthesis of favipiravir. Chin J Pharm 44(9):841–844
Zhang T, Zhai M, Ji J, Zhang J, Tian Y, Liu X (2017) Recent progress on the treatment of Ebola virus disease with Favipiravir and other related strategies. Bioorg Med Chem Lett 27:2364–2368. doi:10.1016/j.bmcl.2017.04.028
Zhao X, Zhou X, Zhong W, Li X (2015) A new antiviral drug-favipiravir. Clin Med J 13(4):16–20
Zheng J, Zhang T, Feng B, Gong X, Kong L, Chen M, Li Y, Niu H, Zhou H, Ding S (2012a) The preparation method of 6-fluoro-3-hydroxypyrazine-2-carboxamide. State Intellectual Property Office of the P.R.C. CN102603658 A (issued July 25, 2012)
Zheng J, Zhang T, Liu C, Feng B, Li Z (2012b) The preparation method of 6-fluoro-3-hydroxypyrazine-2-carboxamide. State Intellectual Property Office of the P.R.C. CN102775358 A (issued November 14, 2012)
Acknowledgements
We are grateful to acknowledge the financial support from the National Natural Science Foundation of China (No. 21302231) and The Modern Analysis and Testing Center of Central South University (CSU).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liu, FL., Li, CQ., Xiang, HY. et al. A practical and step-economic route to Favipiravir. Chem. Pap. 71, 2153–2158 (2017). https://doi.org/10.1007/s11696-017-0208-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-017-0208-6