
Abstract
Low-grade bauxite ores are not favorable in the conventional Bayer process for alumina production, as they are producing more bauxite residue (red mud) and accompanying lower alumina yield than high-grade ores. In the current study, the thermodynamics and characterization of calcium-aluminate slags and pig iron produced from smelting reduction of high iron- and silica-containing bauxites are studied. Coke and limestone are used to reduce the iron oxide and adjust the basicity of slag during smelting. There is evidence that complete iron separation from bauxite is feasible through smelting-reduction process, and up to 99.9 pct of iron can be eliminated. Moreover, it is shown that the partial separation of silicon, titanium, and other elements from the Al2O3-containing slag occurs. The phase compositions and the distribution of elements between the metal and slag phases provide information about the high-temperature behavior of the bauxite components during smelting reduction. Employing electron microscopy analysis, it is indicated that the morphologies of CaO·Al2O3, 12CaO·7Al2O3, 2CaO·Al2O3·SiO2, and CaO·Al2O3·SiO2 phases in the slag, as well as the complex oxides of Ca-Al-Si-Ti in the slag behave differently as the mass ratio of Al2O3/(Fe2O3 + SiO2) in the bauxite changes. It is also shown that the phases of slag produced from smelting-reduction below 5 K s−1 of cooling rate are proper for further leaching process.
Similar content being viewed by others


Background
Data from alumina processing industry show an estimated global average production of bauxite residue (red mud) to be 1 to 1.5 ton per ton of alumina, or about 150 million ton of red mud is produced annually.[1] A large number of efforts have been made to valorize this most-abundant industrial byproduct in the world in building materials,[2] using it as the rare earth element’s source,[3] producing green direct-reduced iron (DRI),[4] consuming inorganic polymers and pozzolanic material,[5] and so on. However, none of these fields has moved to a large-scale commercial production due to economic reasons and particular challenges in processing the red mud. Thus, this caustic material is a prominent environmental issue and is a major concern on most of alumina refineries in the world, and obviously, increasingly lower grade of bauxite (high iron and silica content) makes the situation even more challenging. In the Bayer process, the iron present in the ore is the main component involved in the production of red mud, while silica causes loss of caustic soda to the solvent due to the high reactivity of the compound. The compositions of the bauxite are important in determining the amount of lime that should be added in the mixtures and, eventually, the ratios of CaO/Al2O3 (C/A) in the slags.
In 1927, Harald Pedersen[6] patented a method to extract alumina from bauxite without yielding any red mud; an overall illustration of the process is shown in Figure 1. This process was run commercially in Høyanger, Norway, during 1928 to 1969 with 17,000 ton of annual production before closing down.[7] During that period, other researchers had adapted the patent for utilizing ferruginous bauxite in Albany,[8] and a pilot-plant of alumina production from non-bauxitic source (clay) in Tennessee.[9] Years later, Miller and Irgens[10] also made preliminary studies on alumina refinery plant with 200,000 tons of annual production, based on the same process. The Pedersen process shows a promising potential for the prevention of red mud production, however, as per to date this process has been abandoned more than four decades. Lack of literature and scientific evidence, especially in smelting-reduction part, has led the current authors to investigate the process thoroughly.

An overview of Pedersen process
Experimental Procedure
This section describes the applied experimental activities about the pyrometallurgical part of the Pedersen process for treating selected low-grade bauxites. Sequentially, the procedure consists of materials preparation and characterization, and the details of smelting-reduction treatment.
Materials Preparation and Characterization
Mixture comprises bauxite ore, lime, and coke was prepared in laboratory scale. Three low grade of bauxite ores, named B-, G-, and I-Bauxites were used in different mixtures. The characteristics of lime and coke based on the provided information are listed in Table I. The amounts of lime and coke used were based on the characteristics of the bauxites, and the lime/bauxite ratios for B-, G-, and I- are 0.55, 0.8, and 0.44, respectively.
Pig iron and slag are the products of the smelting-reduction treatment. For the targeted slag compositions, the CaO/Al2O3 (C/A) mass ratios were in the range of 0.65 to 0.75. This C/A ratio range is appropriate to obtain slag phases that has good leaching properties, as was reported in the literature.[11,12] The amounts of coke in the current study are 1.5 times larger than the stoichiometric needs to assure the complete reduction of iron oxides. Coke was dried in an oven at 373 K (100 °C) for 12 hours before use to remove any free-moisture content.
Parts of the ores and products were ground by ring mill into powder size for phase identification using Bruker D8 A25 DaVinciTM X-ray Diffraction (XRD) with CuKα radiation, 10 to 75 deg diffraction angle, 0.01 deg step size, and 2.5 deg for both primary and secondary soller slits. The slag and pig iron samples were mounted in epoxy, polished, and carbon coated before employing Hitachi SU6600TM Scanning Electron Microscope (SEM) to capture the secondary electron (SE) and backscattered electron (BSE) images, X-ray element mapping, and use of Energy Dispersive Spectroscopy (EDS). X-ray Fluorescence (XRF) analysis was used to identify the overall composition of the ores and pig iron and slag products. Moreover, a JXA-8500FTM Electron Probe Micro-Analyzer (EPMA), supported by Wavelength Dispersive Spectroscopy (WDS), was applied for achieving high accuracy on quantitative analysis of elements in selected phases of the products.
Smelting-Reduction Treatment
The mixture of starting materials was set into an open graphite crucible with inner diameter of 115 mm. The crucible was placed in a 75-kVA induction furnace and heated slowly at the rate of 5 K s−1 until it reached 1923 K (1650 °C). The partial pressure of gas (O2, N2, CO, CO2, etc.) inside of the crucible was not measured during the smelting trial. However, an oxidizing atmosphere during smelting reduction process can be considered to exist because the experiments were in the open furnace. The smelting-reduction duration was one hour at 1923 K (1650 °C), while the top of crucible was partially closed by refractories to minimize dissipation of the heat. However, the top was opened at intervals to crush the sintered materials or a solidified foamy-slag, while stirring of the melts was proper due to the induction in pig iron and gas bubbles movements in the slag. To compensate the heat loss, the mixture was heated up to 2023 K (1750 °C) during the last ten minutes of the treatment. Consequently, the means and standard deviations of B-, G-, and I-Bauxite smelting temperatures are 1937 ± 44 K, 1935 ± 54 K, and 1910 ± 38 K, respectively.
The crucible was then cooled to the room temperature inside the furnace, while the power was off. To measure the temperature during the treatment, a tungsten/rhenium thermocouple (type C) inside a graphite rod, and an encapsulating alumina insulation tube were used. About two-thirds of the materials smelted in the first one hour of treatment, and after it cooled down to room temperature, the rest of materials were added, and another one hour of treatment was performed. For further analysis, the solidified materials and the crucible were crushed to separate slag and metal from the crucible.
Results
Results comprise four major parts: analysis of bauxites, smelting-reduction treatment, overall chemical compositions, and phase analysis in products.
Characteristics of Bauxites
An XRD analysis of the bauxites is presented in Figure 2. The mineralogy of B-Bauxite comprises kaolinite, gibbsite, cristobalite, goethite, anatase, and magnetite. The first three are shown as the dominant phases. G-Bauxite predominant phases are diaspore, boehmite, calcite, and hematite. It also has anatase as one of the gangue mineral. I-Bauxite is high in kaolinite and hematite, with relatively low amounts of diaspore and boehmite compared with G-Bauxite. It is worth noting that I-Bauxite has the highest intensity of anatase peaks compared with the other two.

XRD analysis of B-, G-, and I-Bauxites
Table II shows the normalized XRF analysis results of B-, G-, and I-Bauxites. Three major elements that contribute to the bauxite leaching extent are aluminum, iron, and silicon. These elements formed as either a hydroxide or oxide in bauxite as seen in XRD analysis presented above. The Loss of Ignition (LOI) content may decompose to air during smelting trial.
Prior to smelting, bauxite is obviously calcined at lower temperatures. Al2O3 and SiO2 in calcined B- and I-Bauxite are formed as Al2O3·2SiO2 which is known as meta-kaolinite or aluminosilicate—a decomposed phase of kaolinite at high temperature. According to Kyriakogona et al.,[13] the degree of decomposition (dehydroxylation) is up to 98 pct when the calcination occurs at 1123 K (850 °C) in 60 minutes. Table III shows the normalized XRF analysis of the calcined bauxites, determined based on both XRF and XRD results.
Smelting-Reduction Behavior
The measurement of cooling rate was carried out ranging from 1873 K to 1573 K (1600 °C to 1300 °C). As seen later in the CaO-Al2O3-SiO2 ternary phase diagram, the latter temperature corresponds with the solidus line, which as expected means no solidification occurs upon cooling to this temperature. The mean measured cooling rates are 4.9, 4.8, and 5 K s−1 for B-, G-, and I-Bauxite, respectively. These cooling rates are well correlated with the masses (slag + iron) in the crucible where higher total mass shows slower cooling rate under similar conditions.
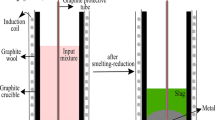
The cross-sectional views of the solidified slag and pig iron in crucible after smelting-reduction trials are displayed in Figure 3. The unreacted coke remaining on top of the slag is also illustrated. The separation of slag and iron produced from B-, G-, and I-Bauxites due to density differences is clearly seen. Obviously, coke particles are floating over the molten slag phase at elevated temperatures and thereafter. The different color appearances of the slags are mostly related to their different compositions. The volume pct values of slag and pig iron produced from different bauxites also were roughly estimated by visual observation. As mentioned in the experimental setup, the order of lime/bauxite mass ratio from the lowest to the highest is as follows: I-Slag, B-Slag, and G-Slag. It appears that the pct of slag increases and that of pig iron decreases considerably with the increasing lime/bauxite mass ratio.

Cross-sectional view of the solidified slag and pig iron produced from B-, G-, and I-Bauxite
In large-scale experiments, a useful performance indicator will be masses of slag and metal (pig iron) produced from a given mass and composition of raw materials. Mass change characteristics, including the correlation between losses of mass with the extent of smelting-reduction reaction, are accordingly of great importance as a reference to large-scale experiments. Table IV shows the changes of mass in different setups after smelting trial. Total mass loss pct values for B-, G-, and I-Setup are 16.3, 15.4, and 6.1 pct, respectively. AQHowever, it becomes more interesting to see only the mass losses of starting materials occurring during reaction and not the losses from both graphite crucible and the rod. As can be seen, the estimated mass losses of materials after smelting trials are considerably high: 41.5, 39.4, and 21.2 pct for B-, G-, and I-Bauxite setups, respectively. Materials’ mass losses after the experiment might be caused by their LOI property, solid–gas transformation, i.e., C → CO(g) or CO2(g), CaCO3 → CaO + CO2(g), and oxide-phase reduction, i.e., FexOy + yC → xFe + yCO(g), SiOy + yC → Si + yCO(g), and TiO2 + 2C → Ti + 2CO(g).
Chemical Compositions of Products
The results of XRF analysis of the produced slags in Table V show the mass balance and smelting of mixtures was done properly as the C/A ratio on each slag is in the expected range, as mentioned in the experimental procedure. It is worth noting here of the low FeOx content in the slag, which agrees with our previous result.[14] AQMost of the iron oxide becomes reduced to metal that saturated with carbon to form pig iron. As seen in Table VI, the B-, G-, and I-Pig iron materials contain 89.9 to 92.7 wt pct Fe and are saturated in carbon, and contain 0.2 to 4.0 wt pct Si, and low traces of titanium and minor elements, i.e., V, Cr, P, and S, are also observed. Most of the minor elements are derived from coke and lime. The saturated carbon content is estimated here based on the silicon content and the reliable solubility data for carbon in Fe-Si-C melts.[15]
Phase and Microstructural Analysis of the Products
Phase identification of the slags resultng from XRD analysis is shown in Figure 4. B-Slag consists of 2CaO·Al2O3·SiO2 (C2AS) and CaO·Al2O3 (CA) as the primary and secondary phases, respectively. However, the latter phase seems to have another polymorph that bonds SiO2 and MgO as is precisely identified later on BSE image. XRD analysis of G-Slag shows that the primary and secondary phases of the slag are composed of 12CaO·7Al2O3 (C12A7), CA and 5CaO·3Al2O3 (C5A3), respectively. The latter phase is considered as an unstable phase that may be reformed to C12A7 phase at high temperature.[16,17] In I-Slag, CAS phase appears as the dominant phase, followed by a small fraction of silica phase.

XRD analysis results on B-, G-, and I-Slag
The X-ray elemental mappings of B-, G-, and I-Slag are shown in Figures 5 through 7, respectively. These figures show clearly the concentration extents of several elements in every structure that coexists in slag. In B-Slag, calcium and silicon are more concentrated in the bright structure, which is later shown to be C2AS phase. Aluminum is more concentrated in the dark area within the lamellar structure. However, titanium and magnesium are dispersed uniformly. In G-Slag, the main structure (C12A7 phase) has more concentration of aluminum than other structures. Silicon and titanium are densely populated between large C12A7 grains. As seen in the figure, there are structures that have complex oxides between Ca-Si-Ti elements. In contrast with B-Slag, magnesium is concentrated in spots. The X-ray elemental mapping of I-Slag shows that titanium, aluminum, and magnesium are highly concentrated in the dark structure. Obviously, bright structure has more concentrations of calcium and silicon than the dark one. Two phases coexist between the C12A7 particles in G-Slag. Therefore, it is necessary to examine the structures in higher magnification and analyze the composition by means of SEM and EDS as the obtained results by these techniques are shown in Figure 8. It shows that the area with bright and dark structures has more concentration of titanium than other structures in slag.

X-ray element mapping of B-Slag

X-ray element mapping of G-Slag

X-ray element mapping of I-Slag

BSE image of G-Slag with the corresponding EDS quantified analysis at (a) through (c) area
Figures 9(a) through (c) show the BSE images of B-, G-, and I-Slag, respectively. As seen in the figure, three structures coexist in the B-Slag and the two main structures are seen in each G- and I-Slags. Composition of each structure was measured by WDS analysis at 2 to 3 locations shown in Figure 9. Table VII presents the WDS analysis of the observed phases in Figure 9. On average, concentrations of CaO, Al2O3, SiO2 constitute more than 95 wt pct of the slags. On the other hand, FeO content is low in all structures of slags, averaging around 300 ppm, which is in agreement with the low measured FeO concentrations by XRF (Table V). Numbers (a)1-3, (a)4-6, and (a)7-9, are WDS results for the dark, fine lamellar, and bright structure that are present in B-Slag (Figure 9(a)), respectively. Dark structure is composed of 43 wt pct CaO, 35 wt pct Al2O3, and 22 wt pct SiO2 on average. Fine lamellar has similar composition as the dark structure has, which is 43 wt pct CaO, 35 wt pct Al2O3, and 21 wt pct SiO2 on average, while bright structure is predominantly composed of 55 wt pct CaO and 31 wt pct TiO2 on average. Numbers (b)1-3 and (b)4-6 denote respectively, the compositions of the bright-coarse grain and smaller grain which are adjacent to a darker structure that is shown in G-Slag (Figure 9(b)). The first structure has 50 wt pct CaO and 49 wt pct Al2O3 on average. The latter one comprises 49 wt pct CaO, 43 wt pct Al2O3, 3 wt pct SiO2, and 4 wt pct TiO2. Numbers (c)1-3 and (c)4-6 show the bright and dark structures in I-Slag (Figure 9(c)), respectively. On average, the bright structure is composed of 31 wt pct CaO, 32 wt pct Al2O3, 34 wt pct SiO2, and 1 wt pct TiO2. The dark structure shows that it has less CaO and more TiO2 than the bright one has: 28 wt pct CaO, 34 wt pct Al2O3, 33 wt pct SiO2, and 3 wt pct TiO2. BSE in I-Slag indicates it has significant number of pores, which are not observed in the B- and G-Slags.

BSE images of (a) B-Slag, (b) G-Slag, and (c) I-Slag
BSE image in Figure 10 shows the cross-sectional views of B-Slag and adjacent B-Pig iron. The light phase shows the Fe-metal of pig iron, while the gray and dark one show carbide and graphite phases, respectively. Table VIII shows the WDS analysis results on the pointed phases. High concentration of titanium is found in carbide phase. Vanadium, as one of the carbide-forming elements, is concentrated in carbide phase, which is shown in points 3 and 4 of Figure 10. Phosphorus and manganese are detected in Fe-metal matrix with 0.2 and 1.1 wt pct on average. Most of the manganese oxide is reduced to pig iron as it has low presence in slags, which is 800 ppm on average. Other trace elements shown in table are recognized as potassium and calcium. In addition, the lower-magnification BSE image in Figure 11 shows the metal matrix and the coexisting phases in the produced metals.

BSE cross-sectional images of B-Slag and adjacent B-Pig iron

BSE images showing the main coexisting phases in (a) B-, (b) G-, and (c) I-Pig iron samples
Discussion
In this section, mass changes before and after of experiment, thermochemistry of the reactions, distribution of elements in slag and pig iron, and slag characteristics are discussed.
Mass Balance
From the LOI data of each bauxite presented in Table II, we can calculate the mass losses in B-, G-, and I-Bauxites due to the ignition, which are 0.28, 0.25, and 0.12 kg for the amounts used, respectively. The LOI data for lime due to the decomposition of lime into CO2(g) can be calculated based on the carbonate amounts in the same table, which are 0.45, 0.66, and 0.23 kg for B-, G-, and I-Bauxite setups, respectively. While the moisture content of coke is removed after preheating in the oven before use, the LOI of coke is 1.53 wt pct as shown in Table I, which gives loss as much as that of 2.10−3 kg.
Total losses due to transformation of C to CO(g) and oxide phase reduction can be calculated and given as 0.16, 0.21, and 0.16 kg for B-, G-, and I-Bauxites, respectively. This gives the theoretical mass losses for B-, G-, and I-Bauxites, respectively, as 0.9, 1.13, and 0.52 kg. As seen in Table IX, the theoretical mass loss with the measured one shows proper correlation, in particular for G- and I-Bauxites. The difference between theoretical and measured mass losses in B-Bauxite might be due to the losses of SiO(g). Details of this explanation will be discussed in Section IV–D.
Smelting-Reduction Thermochemistry
Based on the XRD results in shown in Figure 2, B- and I-Bauxites are constructed mainly from kaolinite. This phase is found in either bauxite or non-bauxitic source, clay. Many works have been carried out to extract alumina from kaolinite with acid or alkaline solution.[18,19,20] In the Bayer process, this is a major problem as the kaolinite is dissolved by sodium hydroxide solution which leads to high loss of caustic soda according to the following reaction:
Furthermore, precipitation of the desilication products, i.e., sodalite (Na8(Al6Si6O24)Cl2), cancrinite (Na6Ca2((CO3)2Al6Si6O24)·2H2O), etc. occur at elevated temperatures that are energy consuming. Low-grade of bauxites are typically characterized by low Al2O3/SiO2 mass ratio[18,21,22] and/or Al2O3/Fe2O3 mass ratio.[23] As both SiO2 and Fe2O3 could be present in particular ore, we may classify the low-grade bauxites based on their low Al2O3/(Fe2O3 + SiO2) mass ratio. According to the concentrations of calcined bauxites in Table III, these ratios for B-, G-, and I-Bauxites are 0.8, 2.4, and 0.6, respectively. These bauxites also have certain amount of TiO2 that later in the slag phase might affect the slag chemistry, its self-disintegration, and leachability.[24] The effect of MgO content of slag on leachability has also been reported as it becomes the main impurity in blast furnaces’ slag.[25] However, in the current study, the compositions of Mg and remaining elements are considered as trace elements, which have minimal effect on the slag properties. According to the obtained XRF results presented in Table V, 94.8 to 99.9 pct of Fe and Mn have been removed from the Al2O3-containing slag, together with parts of Si and Ti. Coke produces CO gas and dissolved carbon in molten iron, and carbon in both forms acts as the reductant for oxides in bauxite. In addition, the usage of graphite crucibles makes an additional carbon source for the carbothermic reduction. Based on the ores’ characteristics, the main smelting-reduction reactions that may take place are as follows:
The underline “_” and parenthesis “( )” notations denote the element or phase that is dissolved in the Fe-liquid solution and slag at known temperature, respectively. It is emphasized here that the reduction of Fe2O3 and Fe3O4 by CO(g) through reactions [3] and [5] is more feasible than reactions [2] and [4]. FeO and even Fe can be also formed in solid state; however, we here assume that Fe is mainly produced from FeO in the slag. Reactions [6] through [10] show the reduction mechanism of FeO and SiO2 at slag/gas, gas/metal, and slag/metal interfaces as observed by Pomfret and Grieveson,[26] Teasdale and Hayes,[27] and Safarian et al.[28] A simple illustration can be given as in Figure 12, where mass transports of the gaseous species (CO, CO2, SiO) in gas bubbles at the slag/metal interface are very important.

Illustration of FeO and SiO2 reduction by carbothermic reaction at slag/gas, gas/metal, and slag/metal interfaces
As lime flux was used for slag making, the main reactions that occur in the process through high-temperature sintering and smelting can be expressed as in reactions [15] through [22]:
Here, we do not consider the FeO-containing phases in the slag, as we do not have much of it left in the final slag. For the sake of simplicity, the compounds that the slag is composed of are denoted as follows: CaO = C, Al2O3 = A, and SiO2 = S. In thermodynamic perspective, the above reactions of pig iron and slag formation and their competition behaviors are dependent on the Gibbs energy changes at particular temperature. The Gibbs energy changes of the formation of the metal and slag components in pure state are calculated using HSCTM software. The reduction of pure iron oxides may occur below 1273 K (1000 °C), while silica and titania are phases that could be reduced by carbon at relatively high temperature, which means the separation of these two phases out of bauxites consumes more energy than iron. Moreover, the reduction of silica by CO (g) is unlikely to occur, as the Gibbs energy changes are positive at any given temperature. The slag-making reactions (reactions [15] through [22]) are viable at low temperature, and there is a large driving force for slag formation upon the heating of bauxite + lime to elevated temperatures. The WDS results in both slag and pig iron (Tables VII and VIII) show that the main phases are in solid solution form with certain solubility, instead of being in the pure forms. However, HSCTM calculations are made for pure substances, and the real Gibbs energy changes for the slag-making reactions may be even more negative than that the calculations indicate.
The equilibrium phases in different mixtures that coexist during and after the smelting-reduction can be calculated using thermodynamic software, FactSageTM, at temperatures between 1273 K and 2073 K (1000 °C and 1800 °C) with an increment of 283 K (10 °C) and 0.70 ± 0.05 of C/A ratios. As the calculation results in Figure 13 indicate, the Fe-fcc phase coexists with the solidified forms of B-, G-, and I-Slags in the temperature range of 1273 K to 1663 K (1000 °C to 1390 °C). The lattice changes to Fe-bcc phase in the range from 1663 K to 1803 K (1390 °C to 1530 °C) before it starts to melt. In the meantime, liquid slag phase appears at around 1473 K (1200 °C), and its amount increases significantly at the range of 1673 K to 1773 K (1400 °C to 1500 °C). B- and I-Slags should have similar solid slag phases that are stable at temperatures below 1573 K (1300 °C) as seen in Figure 13. The sequential order from the highest to the lowest fraction of phase is CAS2 > C2AS > CaTiO3 > CaMg2Al16O27 > Ca2Mg2Al28O46 > spinel. In addition, B-Slag has CaAl12O19 phase as well at 1673 K (1400 °C). The calculation results of the coexisting equilibrium phases in B- and I-Slag are not in agreement with the XRD results presented in Section III. The reason of the disagreement might be related to the element’s distribution during smelting-reduction trial that will be discussed in Section IV–E.

The calculated phases in equilibria at different temperatures for (a) B-Bauxite, (b) G-Bauxite, and (c) I-Bauxite setups
G-Slag has CA phase that constitutes about 70 wt pct of the slag phase, followed by low amounts of C2AS, Ca3MgAl4O10, Ca2SiO4, Ca3Ti2O7, spinel, and KAlO2. FactSageTM does not recognize C12A7 phase in its database, and it is the reason that this phase is absent in the equilibrium phases of G-Slag. Apart from C12A7 phase, the presence of CA phase is in agreement with the XRD result of G-Slag. It is worthy to note that the amounts of TiO2 and MgO of all obtained slags as in Table V are considerably low, which causes to not observe CaTiO3, CaMg2Al16O27, etc. in XRD analysis. However, the thermodynamic calculations here show that such phases may coexist with the main recognized phases in the slags.
Effect of Slag Viscosity on Porosity
The significant number of pores that appear in I-Slag in Figure 9(c) give strong indication of the gas formation during the oxides’ reduction by the mechanism illustrated in Figure 12. The pores have been formed from the trapped gas that might emerge from the reduction reactions at the slag/metal interface. I-Slag has the highest amount of SiO2 compared to others, and the basicity based on CaO/SiO2 (C/S) mass ratio is 0.9, which is relatively acidic. This slag forms networking oxides in melts, which increase the viscosity and hinder the gas movement. In order to clarify this, FactSageTM was applied to calculate the viscosities of the obtained B-, G-, and I-Slags produced at 1923 K (1650 °C) and viscosities of 0.17, 0.10, and 0.48 Pa s were, respectively, obtained as shown in Figure 14. The figure shows the correlation between different C/S mass ratios with the viscosities at 1723 K, 1823 K, and 1923 K (1450 °C, 1550 °C, and 1650 °C), where C/A mass ratio is equal to 1. It can be seen that by decreasing the temperature, the viscosity is significantly increased especially at the C/S mass ratio of less than 3. That is to say, the gas bubbles forming and leaving the slag/metal interface have more difficulties to rise in I-Slag than in the others as the I-Slag has more gas storage capacity. It is worth mentioning that during the smelting-reduction experiments more foaming of the I-Slag was observed than for the others, which is supporting the viscosity effect.

Correlation between basicity (C/S) and viscosity in different C-A-S slags at different temperatures
Silica Losses from Slags
The theoretical and measured mass loss differences in G- and I-Bauxites are relatively minor, whereas B-Bauxite shows otherwise (Table IX). This indicates that another factor has contributed to the loss of mass in B-Bauxite that might be elaborated as follows.
According to the XRF analysis of bauxite, B- and I-Bauxites have identical concentration of SiO2. It is worthy to note that both have kaolinite as their main phase. Based on material balance calculation, total amounts of silicon either in starting materials and the products produced from B- and I-Bauxites should be 0.29 and 0.17 kg, respectively. In fact, both of the bauxites show nonequivalent amounts of silicon in their input–output balance. The estimated losses of silicon in B- and I-Slags are 72.7 and 17.6 pct, respectively.
Silicon could be lost as a gas phase SiO(g), which is produced from reaction between SiO2 in slag with carbon in the metal, as in reaction [23]. Then, it is exiting from the slag when the gas bubble rises to the surface.
The Gibbs energy change of formation in reaction [23] at 2023 K (1750 °C) is − 2.2 kJ, which shows that high temperature is an important factor in the silica gas production from the slag. As mentioned in the experimental procedure, the mean smelting temperature of B-Bauxite is higher than that of I-Bauxite. Silica loss could explain one of the reasons why the concentration of silica in B-Slag is lower compared with I-Slag, although the mixture setup before the smelting trial has identical C/(A + S) mass ratios, that is, 0.36 and 0.34 for B- and I-Bauxites, respectively.
The Distribution of Elements Between Slag and Pig Iron
Based on equilibrium calculations in Figure 13, the main phases of B- and I-slags at low temperature should be CAS2 and C2AS phase, respectively. In contrast, the XRD results in Figure 4 show the main phases for B- and I-Slags to be C2AS and CAS, respectively. This result is supported by XRF analysis of the products, which shows different compositions in slag and pig iron produced from B- and I-Bauxites, regardless of the similar phase and C/A ratio that the bauxite mixtures have before. This gives indication that during smelting-reduction and solidification of the melt, the elemental distribution may act as a decisive factor on the final phases in slag.
Figure 15 shows the distribution of phases in calcined bauxite ores and the corresponding slags after smelting-reduction according to the XRF results. The weight fractions of Al2O3 and SiO2 phases remain the same before and after the treatment, which indicates that the two phases are not easily reduced during the smelting-reduction treatment. The significant decrease in the quantity of SiO2 phase in B-Slag has already been discussed in Section IV–D. The CaO wt pct in G-Slag is higher than that in other slags because of the high amount of lime used to maintain the required C/A ratio to achieve the leachable calcium-aluminate phase.

Distribution of phases in bauxite ores and slags after smelting-reduction
The reduction extent of each phase can be calculated by simple mathematical expression:
where W° is mass of the amount of metal based on the stoichiometry, as if 100 pct was reduced from an oxide and W is the actual amount of that metal present in the metal phase based on the XRF result of the pig iron. To simplify the mass loss based on the stoichiometry principle, we assume the iron reduction is 100 pct as the average WDS results of FeO content in all slags are low (see Table VII). Therefore, the SiO2 reduction extents in B-, G-, and I-slag are 2.8, 1.3, and 1.6 pct, respectively. Higher SiO2 reduction in B-Bauxite might be due to the higher smelting temperature of this trial compared with G- and I-Bauxites as discussed above. According to HSC data, reaction [8] transposes to the right side at temperature more than 1938 K (1665 °C), and based on the smelting temperature setup described in Section II–B, B-Bauxite was exposed to this temperature relatively longer than others. From kinetics point of view, carbothermic reduction of SiO2 from slag is much slower than that of FeO. The other partially reduced phase was TiO2. The TiO2 reduction extents are 2.8, 8.4, 1.5 pct for B-, G-, and I-Bauxites, respectively. This calculation is in agreement with the amount of TiC that is observed in BSE image of the pig iron shown in Figure 11. The reduction of TiO2 in reaction [11] proceeds at around 2023 K (1750 °C) when pure substances are involved. However, as the activity of titanium in the molten iron is low, the reduction of TiO2 may occur at the lower process temperature to some extent, yielding dissolved titanium in iron. Under carbon-saturated condition, the dissolved titanium has high possibility to make bond with the existing carbon and forms TiC during solidification of pig iron (reaction [25]). This is further enhanced due to the low solubility of titanium in Fe-Csaturated-Ti system.[29]
Another mechanism for the formation of TiC particles can be the direct reaction of the dissolved carbon in the melt with TiO2 in the slag at slag/metal interface, as in reaction [26].
The thermodynamic activities given in reactions [11] and [26] are performed under standard conditions with the titanium activity being unity. Therefore, we need to evaluate the reactions under non-standard conditions, where we have a low titanium activity in the melt. For reactions [11] and [26], the expression of Gibbs energy changes can be written as
where ΔG11° and ΔG26° are the standard Gibbs energy changes of reactions [11] and [26], respectively. The activities of C and TiO2 in the melt is unity and unknown, respectively. The partial pressure of CO(g) for both reactions is the same, thus Eqs. [27] and [28] can be rearranged to
Since activity of TiC is unity, then Eq. [30] becomes
By comparing δG11 and δG26 values, we may predict which reaction is more favorable to occur at a particular temperature. We determine the activity of titanium in Fe-Csaturated-Ti system at 1873 K (1600 °C) from FactSage™ database and calculate the δG11 and δG26 in the melt, as seen in Figure 16.

The changes of δG11 and δG26 at 1873 K in different mol pct Ti
Figure 16 shows that if the amount of titanium in the melt is less than about 0.01 mol pct, the reaction [11] is more favorable than reaction [26], which means the TiO2 is reduced and yields the dissolved titanium in the metal. On the contrary, if the titanium content is larger than about 0.01 mol pct, then reaction [26] is more favorable than that of reaction [11], which means TiC formation at slag/metal interface is likely to occur.
As seen in WDS analysis of pig iron in Table VIII, silicon content on numbers 1 and 2 of B-Pig iron show higher amounts compared with that of silicon content in I-Pig iron (Table VI). This confirms the result in XRF analysis, and demonstrate the reduction mechanism of silica in bauxite during smelting-reduction. The WDS results of point numbers 3 and 4 shown in Table VIII confirmed the formation of (Ti,V)C in metal as was reported from the literature.[7] As the solidus temperature of metal is lower than that of slag, the solute titanium in pig iron may segregate to the boundaries of metal and slag, and forms carbide.
Another important parameter in studying the slag-metal systems is the distribution of the dissolved elements between the slag and metal phases, and a distribution coefficient as Li is defined, which is expressed as
where (wt pct i) and [wt pct i] represent the mass pct of element i in the slag and metal phases, respectively. The distribution coefficients of Fe, Si, Ti, Mn, P, and V in the current work are shown in Figure 17. Fe, Mn, P, and V have relatively low LFe, LMn, LP, and LV values (0 to 1.7), which indicates that these elements are stable in pig iron and shows that iron was 99.9 pct reduced. On the other hand, the LSi and LTi values vary depending on the type of bauxite and so the slag compositions, which are 1.3 to 11.6 and 0.8 to 8.2 for LSi and LTi, respectively. More experiments are necessary to obtain better understanding on the elemental distribution in slag and pig iron produced from smelted bauxite.

Distribution coefficients of several elements in different bauxites
Stable Phases in the Slag System
Even though the mixtures were set up with particular ratio of C/A, the other compounds, i.e., SiO2 and TiO2 affect the construction of phases in the slag system. AQAs seen in Table V, the main slag compounds are CaO, Al2O3, and SiO2, while TiO2 content is negligible (< 3 wt pct) and is not forming any new phase in the system.[24] Therefore, the slag is regarded here as a ternary CaO-Al2O3-SiO2 system. Employing FactSage™ phase diagram module, the projection of isothermal sections from 1573 K to 1873 K (1300 °C to 1600 °C) in CaO-Al2O3-SiO2 system can be made as shown in Figure 18. Due to the limitation of compound database in FactSage, C12A7 phase is not shown in the projection. However, we draw the area of C12A7 phase manually into the ternary system (colored in pink) based on the provided information in the literature.[17,30]

Isothermal projection of ternary CaO-Al2O3-SiO2 system
The calculated ternary phase diagram corresponds with the XRD result of B-Slag in Figure 4. The direction of crystallization path proceeds toward the peritectic temperature and shows that C2AS, CA, and β-C2S phases are in equilibrium as condensed phases after solidification. However, neither β-C2S nor γ-C2S phases can be traced in the diagram. The absence of C2S may be the reason why self-disintegration did not happen during cooling, as we observed. The self-disintegration of slag occurs due to the volume expansion by 12 pct as the crystalline phase transforms from β-C2S to γ-C2S.[24,31] In calcium-aluminate-containing-slag produced from blast furnace, it is reported that when the cooling rate is lower than 4.8 K s−1, then the major phases of the slag are C12A7, γ-C2S, and CA.[32] On the other hand, if the cooling rate is considerably high, that is, 4.8 to 5.4 K s−1, the formation of C2AS occurs. In the current study, the cooling rate averages were above 4.8 K s−1. Another possible explanation would be related to the low presence of β-C2S in B-Slag (< 5 pct), as calculated in Section IV–G, which is confirmed by no observation of the corresponding peaks in XRD spectra (Figure 4).
Based on the XRD results, G-Slag has C12A7, CA, and a low fraction of C5A3 phase. The C12A7 phase was first observed by Rankin and Wright[33] in 1915 and has been discussed several times ever since. In 1965, Nurse et al.[34] observed that C12A7 phase is unstable in anhydrous or moist-free CaO-Al2O3 system, as it is in equilibrium with H2O above 1223 K (950 °C) and forms Ca12Al14O32(OH)2 when is fully saturated: the water is present as hydroxyl ions in crystal structure. Later on, Haccuria et al.[35] showed that in a dry and high-purity (99.999 pct) argon atmosphere, the formation of C12A7 phase does not occur. Here in G-Slag, C12A7 phase is observed in its anhydrous form with the data given as follows: a = 11.99856 Å, V = 1727.38 Å3, and density = 3.10−3 kg m−3, in cubic I-43d (220) space group. The phase in the slag samples have been formed in an open furnace and oxidizing atmosphere, which subsequently is cooled to room temperature. The result is in agreement with Imlach et al.[36] who hypothesized that the C12A7 phase might be stable at high temperatures as it absorbs the excess of oxygen from any oxidizing atmosphere, even where pO2 is as low as 10−3 Pa (10−8 bar). This finding is important to the study of calcium-aluminate slag making, as C12A7 phase is known as one of the leachable phases in CaO-Al2O3 system[8,11,17,37] and is, therefore, crucial to Pedersen process. Hallstedt[16] optimized the thermodynamic data of C12A7 phase and proposed that it melts congruently at 1449 °C. It has a eutectic with other phases C3A and CA on each side, as shown in Figure 19(a). In our results, G-Slag consists mainly of 46.1 wt pct Al2O3 and 48.1 wt pct CaO, which means according to Figure 19(a) it may rather have C3A and C12A7 than C12A7 and CA phases. AQHowever, Hallstedt considers only the binary system of CaO-Al2O3 and not the role of other constituents even if they were involved, such as silica, titania, etc. Moreover, C3A phase is relatively unstable as it can be reacted with free-Al2O3 in the system to form C12A7 (see reaction [18]). In our previous study,[12] we did not observe the presence of C12A7 phase in a slag that was synthesized at high temperature and under oxidizing atmosphere. The composition was 38 wt pct and 62 wt pct of CaO and Al2O3, respectively. Instead of the occurrence of CA and C12A7 phase, the encountered phases were CA and C3A, and this result agrees with a moisture-free binary diagram as shown in Figure 19(b).

Part of CaO-Al2O3 binary system where (a) the C12A7 phase is included and (b) a moisture-free without formation of C12A7 phase, as simulated by FactSageTM
The reason of this different phenomenon can be explained as follows:
-
1.
In our previous study,[12] CA phase was the primary crystallized phase (91 wt pct by phase lever-rule), and when the cooling reached eutectic temperature, the remaining liquid may precipitate the C12A7 phase at grain boundaries. However, the amount of this phase was low (< 10 wt pct), and either it was dissolved in the adjacent CA phase or is dissociated to CA and C3A phases at lower temperature as shown in reaction [33].
$$ 2 {\text{C}}_{ 1 2} {\text{A}}_{ 7} = {\text{ 5C}}_{ 3} {\text{A }} + {\text{ 9CA}} $$(33)As reported in the literature,[34] the Gibbs energy changes for reaction [33] are − 1.5, − 2.0, and − 3.5 kJ/mole CaO at 1700 K, 1600 K, and 1500 K (1427 °C, 1327 °C, and 1227 °C), respectively. Therefore, formation of C3A and CA phases upon cooling was favorable.
-
2.
In the current study, as indicated by the strongest intensity in XRD in Figure 4, the primary crystallized phase from G-Slag is C12A7. As it has significant amount, it may absorb the required oxygen during precipitation and eventually become more stable. Based on this reasoning, we may hypothesize that unless the composition of the binary slag is on the range of its primary crystallization (about 46 to 51 wt pct Al2O3) or as the majority phase in the system, the C12A7 phase is likely to be unstable even in oxidizing atmosphere.
According to the XRD results in Figure 4, no CAS2 phase in I-Slag is observed, which does not agree with the ternary system at 101.3 kPa (1 bar) shown in Figure 18. CAS that is observed is known as Calcium Tschermak’s pyroxene (CATs) for geologists, which is one of the important constituent of the earth’s upper mantle and of the natural aluminous pyroxenes. The crystallography pattern of CAS in the current study matched against the measurement of Okamura et al.[38]: a = 9.609 Å, b = 8.652 Å, c = 5.274 Å, β = 106.06 deg, V = 421.35 Å3, in monoclinic C2/c space group. The CAS phase is formed by a solid-state reaction as seen in reactions [34] and [35]. Okamura et al.[38] synthesized a single crystal of CAS from CAS2, C2AS, and Al2O3 after heating the mixture at 1573 K (1300 °C) within 24 hours in 18.105 kPa (18 kbar). On the other hand, Hariya and Kennedy[39] observed that CAS2 decomposes to CAS and SiO2 under high pressure and temperature.
Many studies concluded that CAS is a high-pressure and temperature phase.[38,40] In addition, Hays[41] reported that pure CAS is not stable below 1433 K (1160 °C), and the pressure field of stability is relatively narrow even at higher temperature. However, Ma et al.[42] stated that although synthetic CAS is a high-pressure phase, a natural CAS could be found in low-pressure atmosphere. Figure 9(c) shows that I-Slag does not have lamellar structure, which indicates the phases were not formed through a slow diffusion mechanism. Thus, in contrast to the conclusion that was given by some researchers above, we suggest that the reaction [35] may occur during the cooling of a congruently melted CAS2 phase at ambient pressure. The coexisting phases of CAS and SiO2 in XRD results of I-Slag are worth as the evidence in support of the argument. Nevertheless, this hypothesis needs further investigation.
Phase Changes upon Slag Solidification and Cooling
In Table VII, the WDS analysis of B-Slag on dark and bright structures is identical with the phases of C2AS and CaTiO3, respectively. The WDS results on fine lamellar phase indicate a C2AS phase as it has similar composition with the dark phase, it only has more TiO2. This may not be precise, as the lamellar is constructed with dark and less-bright phase. It is difficult to identify the composition of each layer as the thickness is as much as 2 µm. To understand the existing phases in lamellar structure, a crystallization path of B-Slag becomes necessary. Based on alkemade line and phase lever-rule we can describe it as follows:
-
1.
Consider we have 100 g of slag with the composition of 50 wt pct Al2O3, 12 wt pct SiO2, and 38 wt pct CaO, which is a normalized XRF data of B-Slag from Table V. At 1923 K (1650 °C), slag liquid phase exists, named liquid #1.
-
2.
From 1923 K to 1783 K (1650 °C to 1510 °C), the primary crystallization of C2AS occurs; liquid #1 → C2AS + liquid #2. The slag consists 14 g of C2AS phase (14 pct) and 86 g of liquid #2 phase (86 pct).
-
3.
From 1783 K to 1623 K (1510 °C to 1350 °C), following reaction occurs: liquid #2 → C2AS + CA + liquid (p). At this stage, lamellar structure coexists. The masses of C2AS, CA, and liquid (p) phase are 30.1 (35 pct), 49 (57 pct), and 6.9 g (8 pct), respectively.
-
4.
At 1623 K (1350 °C), liquid (p) + C2AS → C2S + CA, a peritectic reaction occurs. At the current stage, the remaining liquid is consumed, as well as the partial of C2AS phase. The masses of liquid (p), C2AS, C2S, and CA phases are 6.9 g (66 pct), 3.55 (34 pct), 3.86 (37 pct), and 6.59 g (63 pct), respectively.
-
5.
From 1623 K (1350 °C) to room temperature, at the solidus line, no liquids exist, and impurities may segregate and remain concentrated between the primary C2AS grains. Total amount of condensed phases in B-Slag is
On the other hand, coarse and small grain structures in G-Slag indicate the C12A7 phase. Grzymek et al.[43] observed that C12A7 has polymorphic transition phase, depending on the synthesizing temperature, condition of atmosphere (reducing or oxidizing), and the cooling rate when it was formed. AQGrzymek mentioned that when synthesis is carried out under an oxidizing atmosphere at 1770 K (1497 °C), and then slowly cooled, the structure is isotropic and grainy-ish with an anisotropic phase formed on grain boundaries, which is similar to the current result. The structure of CA phase in G-Slag is indistinguishable as the amount is considerably low.
In I-Slag, pointed WDS analysis on bright and dark structures correspond to CAS phase with insignificant difference on TiO2 amount (1 to 2 wt pct). No lamellar structure is observed as it was in B-Slag. An X-ray elemental mapping on I-Slag in Figure 7 shows the dark structure has high concentration in titanium, and it has stronger bonding affinity to aluminum than that of calcium and silicon. Interestingly, these complex oxides do not behave similarly in G-Slag. The areas of the phase that is rich in titanium have tendency to preferentially bond with calcium and silicon rather than aluminum, as shown in Figure 6, which is supported by SEM and EDS results for G-Slag in Figure 8. This gives valuable information for the leachability of the slag, as the titanium that bonded to the aluminum might hinder the alumina-leaching efficacy.
As we observed above, the slags B, G, and I contain different Al2O3-containing phases, and the leachability of them is important for extraction of alumina. The leaching behavior of C12A7 and CA phase has been studied in the literature, and the main point to be mentioned here is that both phases are soluble in sodium carbonate solution and therefore is favorable for the alumina recovery. Lundquist and Leitch[44] proposed the leaching reaction of CA phase where
while for C12A7 phase, the reaction may be described as follows[8]:
However, no publication was found about the leachability behaviors of CAS, C2AS, and C5A3 phases. This will be the subject of study of the current authors in the near future. Understanding the leaching behaviors of Al2O3-containing phases may pave the way to treat different metallurgical slags, which is quite important regarding the sustainable process development and circular economy.
Conclusion
The characteristics of calcium-aluminate-containing slags and pig iron produced from low-grade bauxite ores were studied. Using advance characterization techniques, the thermochemistry of oxides reduction, elemental distributions between the slag and metal products, and mechanisms of the formation of phases in them can be explained. The main conclusions drawn can be summarized as follows:
-
1.
Iron from the ores is removed from 94.8 to 99.9 pct during smelting-reduction by carbon and yields the alumina-containing slag containing less than 1 wt pct Fe, dependent on the slag chemistry.
-
2.
The partial separation of Mn, Si, Ti, V, and P oxides occurs through the smelting-reduction, where low distribution coefficients of LMn, LP, and LV are observed, while LSi and LTi values are in ranges that are considerably wide depending on the characteristics of the produced slag.
-
3.
The viscosity in calcium-aluminate slags with CaO/SiO2 mass ratio less than three is higher compared with other compositions at elevated temperatures. It causes lower Fe-removal and yields more porous product.
-
4.
Silicon loss from high SiO2-containing bauxites may occur at elevated temperatures through the SiO gas formation.
-
5.
The leachable slag phases (CaO·Al2O3, 12CaO·7Al2O3), which are observed in G-Slag, are obtained after the smelting-reduction of bauxite that has Al2O3/(Fe2O3 + SiO2) mass ratio higher than two at cooling rate of up to 5 K s−1.
-
6.
The complex bonding between titanium and aluminum oxides in slag is not favorable for leaching process and is likely to occur in CaO·Al2O3·SiO2 phase.
References
K. Tsesmelis: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, p. 71.
D.J. Roth and J. Falter: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, pp. 539–45.
E. Ujaczki, P. Cusack, S. Clifford, T. Curtin, R. Courtney, and L. O’Donoghue: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, pp. 485–90.
B. Bhoi, P. Rajput, and C.R. Mishra: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, pp. 565–74.
T. Hertel, B. Blanpain, and Y. Pontikes: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, pp. 575–85.
H. Pedersen: United States of America Patent, 1927, 1618105.
J. Safarian and L. Kolbeinsen: Sustainable Industrial Processing Summit, 2016, pp. 149–58.
H.E. Blake Jr., O.C. Fursman, and A.D. Fugate: US Bureau of Mines, 1966, pp. 1–19.
T. Hignett: Industrial and Engineering Chemistry, 1947, vol. 39, pp. 1052-60.
J. Miller and A. Irgens: Light Metals, 1974, Springer, Cham, pp. 977-82.
R. Lundquist and H. Leitch: US Bureau of Mines, 1964, pp. 1–16.
F.I. Azof, L. Kolbeinsen, and J. Safarian: Proceedings of 35th International ICSOBA Conference, 2017, vol. 42, pp. 243–53.
K. Kyriakogona, I. Giannopoulou, and D. Panias: Proceedings of the 3rd World Congress on Mechanical, Chemical, and Material Engineering, 2017, pp. 1331–36.
H. Sellæg, L. Kolbeinsen and J. Safarian: Light Metals, 2017, Springer, Cham, pp. 127-135.
S. H. Ahn, Y. H. Kim, J. P. Shin and Y. E. Lee: ISIJ International, 2014, vol. 54, pp. 750-55.
B. Hallstedt: Journal of the American Ceramic Society, 1990, vol. 73, pp. 15-23.
K.-S. Chou: Retrospective theses and dissertations, Digital Repository of Iowa State University, 1979, pp. 1–152.
A. Pehlivan, A.O. Aydin, and A. Alp: Sakarya University Journal of Science, 2012, pp. 92–98.
A. W. A. Al-Ajeel and S. I. Al-Sindy: Iraqi Bulletin of Geology and Mining, 2006, vol. 2, pp. 67-76.
J. Xiao, F. Li, Q. Zhong, H. Bao, B. Wang, J. Huang and Y. Zhang: Hydrometallurgy, 2015, vol. 155, pp. 118-124.
P. Smith: Hydrometallurgy, 2009, vol. 98, pp. 162-176.
M. Jiayu, L. Zhibao and X. Qinggui: American Institute of Chemical Engineers, 2012, vol. 58, pp. 2180-91.
H.E. Blake Jr., O.C. Fursman, A.D. Fugate, and L.H. Banning: US Bureau of Mines, 1968, pp. 1–19.
Z. Tong, Y. Li and L. Lian: Light Metals, 2012, Springer, Cham, pp. 185-88.
B. Wang, H.-L. Sun, D. Guo and S.-W. Bi: Light Metals, 2011, Springer, Cham, pp. 201-4.
R. Pomfret and P. Grieveson: Canadian Metallurgical Quarterly, vol. 22, pp. 287-299, 1983.
S. Teasdale and P. Hayes: ISIJ International, 2005, vol. 45, pp. 634-41.
J. Safarian, L. Kolbeinsen, M. Tangstad and G. Tranell: Metallurgical and Materials Transactions B, 2009, vol. 40B, pp. 929-39.
M. Sumito, N. Tsuchiya and K. Okabe: Transactions ISIJ, 1981, vol. 21, pp. 414-21.
M. Kowalski, P. Spencer and D. Neuschutz: Slag atlas 2nd edition, Dusseldorf: Verlag Stahleisen GmbH, 1995.
H.-L. Sun, B. Wang, H.-Y. Yu, S.-W. Bi and G.-F. Tu: Light Metals, 2010, Springer, Cham, pp. 29-32.
B. Wang, H.-L. Sun, X.-Z. Zhang and S.-W. Bi: Light Metals, 2011, Springer, Cham, pp. 241-44.
G. Rankin and F. Wright: American Journal Science, 1915, vol. 39, pp. 1-79.
R. Nurse, J. Welch and A. Majumdar: Transaction of British Ceramics Society, 1965, vol. 64, pp. 323-32.
E. Haccuria, T. Crivits, P. C. Hayes and E. Jak: American Ceramic Society, 2015, vol. 99, pp. 691-704.
J. Imlach, L. Glasser and F. Glasser, Cement and Concrete Research, 1971, vol. 1, pp. 57-61.
H.-L. Sun, B. Wang, J.-X. Zhang, and S.-F. Zong: Adv. Mater. Sci. Eng., 2014, pp. 1–6.
F. P. Okamura, S. Ghose and H. Ohashi: American Mineralogist, 1974, vol. 59, pp. 549-57.
Y. Hariya and G. Kennedy: American Journal of Science, 1968, vol. 266, pp. 193-203.
B. J. Wood: American Mineralogist, 1976, vol. 61, pp. 599-602.
J.F. Hays: Carnegie Institution Washington Year Book, 1965, pp. 234–39.
C. Ma, S. B. Simon, G. R. Rossman and L. Grossman: American Mineralogist, 2009, vol. 94, pp. 1483-92.
J. Grzymek, A. Grzymek, Z. Konik, A. Stok, and M. Gawlicki: Light Metals, 1985, Springer, Cham, pp. 87-99.
R. V. Lundquist and H. Leitch: US Bureau of Mines, 1963, pp. 1–9.
Acknowledgments
The authors thank Hanne Sellæg for her valuable contribution in the laboratory experiments. The current research has been funded by the NTNU and cosponsored by the Research Domain 5–Materials and the Society in SFI Metal Production (a Norwegian Centre for Research-driven Innovation in metal production) under the Project Number 237738.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted February 12, 2018.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Azof, F.I., Kolbeinsen, L. & Safarian, J. Characteristics of Calcium-Aluminate Slags and Pig Iron Produced from Smelting-Reduction of Low-Grade Bauxites. Metall Mater Trans B 49, 2400–2420 (2018). https://doi.org/10.1007/s11663-018-1353-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-018-1353-1