
Abstract
Background
Genotype-guided initial warfarin dosing may reduce over-anticoagulation and serious bleeding compared to a one-dose-fits-all dosing method.
Objective
The objective of this review was to investigate the safety and efficacy of genotype-guided dosing of warfarin in reducing the occurrence of serious bleeding events and over-anticoagulation.
Data Sources
The authors searched PubMed, EMBASE and International Pharmaceutical Abstracts through January 23, 2009, without language restrictions. Selected articles were randomized trials comparing pharmacogenetic dosing of warfarin versus a “standard” dose control algorithm in adult patients taking warfarin for the first time.
Review Methods
Two reviewers independently extracted data and assessed study quality using a validated instrument. The primary outcomes were major bleeding and time spent within the therapeutic range International Normalized Ratio (INR). Secondary outcomes included minor bleeding, thrombotic events and other measures of anticoagulation quality.
Results
Three of 2,014 studies (423 patients) met the inclusion and exclusion criteria. Differences in study quality, dosing algorithms, length of follow-up and outcome measures limited meta-analysis. Summary estimates revealed no statistically significant difference in bleeding rates or time within the therapeutic range INR. The highest quality study found no significant difference in primary or secondary outcomes, although there was a trend towards more rapid achievement of a stable dose (14.1 vs. 19.6 days, p = 0.07) in the pharmocogenetic arm.
Conclusions
We did not find sufficient evidence to support the use of pharmacogenetics to guide warfarin therapy. Additional clinical trials are needed to define the optimal approach to use warfarin pharmacogenetics in clinical practice.
Similar content being viewed by others


BACKGROUND
In 2007 the Food and Drug Administration (FDA) issued a labeling change advising physicians to consider the use of “genetic tests to improve their initial estimate” of warfarin dose.1 This is the first FDA recommendation to consider genetic testing when initiating a commonly prescribed medication and may set a precedent for the future use of genetic technologies in clinical practice. Many forces are driving this technology into practice with increasing numbers of companies promoting testing,2–4 academic institutions racing to be on the cutting edge of clinical medicine, and patients5 interested in the potential of personalized medicine. Indeed, warfarin has several attributes that make it an attractive “target” of personalized medicine: it is commonly prescribed,6 has a narrow therapeutic index with up to 20-fold inter-individual variability in dose-response,7,8 an annual major bleeding risk of 1–5%9–13 and several common genetic variants that affect warfarin metabolism and activity.14–16
Warfarin dose variability is associated with many factors, including age, height, body weight, race, dietary vitamin K intake, intercurrent illness, drug interactions and genetic variation.17,18 Among Caucasians, an estimated 30% to 40% of warfarin dose variability can be attributed to polymorphisms in genes encoding hepatic isoenzyme cytochrome P-450 2C9 (CYP2C9), which is responsible for metabolic clearance of warfarin, and vitamin K epoxide reductase complex subunit 1 (VKORC1), the enzymatic target of warfarin.8,14,15,19–28
Allelic variants in CYP2C9 and VKORC1 are common, with more than two thirds of the Caucasian population and up to 90% of East Asians manifesting at least one variant.14–16 Affected individuals require, on average, lower doses of warfarin to maintain a therapeutic INR and more time to achieve stable dosing.15,19,21,23,29–31 Carriers of variant alleles are at higher risk for bleeding complications, particularly at the induction of warfarin therapy,32–37 and genotype-guided dosing algorithms better approximate maintenance warfarin dose than fixed-dose algorithms.15,32,38–43 However, a recent analysis by Eckman and colleagues concluded that genotype-guided dosing was unlikely to be cost-effective in nonvalvular atrial fibrillation patients.44 Furthermore, it remains unclear whether pharmacogenetic dosing will reduce the incidence of serious bleeding or over-anticoagulation compared to current methods of initiating and dose-adjusting warfarin.
OBJECTIVES
In order to summarize the current evidence supporting the use of warfarin pharmacogenetics, we performed a systematic review of randomized trials that compared a dose-selection strategy that used pharmacogenetic information to one that did not.
METHODS
Data Sources
We searched PubMed, EMBASE and the International Pharmaceutical Abstracts through January 23, 2009. The complete search strategy is described in the online Appendix 1. In order to identify ongoing clinical trials, we searched http://www.clinicaltrials.gov on February 19, 2009 (online Appendix 1). We examined the reference lists of included articles and professional reviews, and contacted experts to identify other potentially relevant studies.
We included randomized controlled trials that compared clinical outcomes among a pharmacogenetic dosing group, using common genetic variants of CYP2C9 and/or VKORC1, to a dosing algorithm that did not incorporate genetic testing. Eligible studies enrolled adult, warfarin-naïve patients with any indication for warfarin therapy, including atrial fibrillation, venous thromboembolic disease, recent orthopedic surgery and valvular disease.
Data Extraction
Two reviewers (KK, JT) independently extracted data using a data abstraction instrument: number of participants in each arm, study quality, length of follow-up period, intervention and control dosing algorithms, and primary and secondary outcomes. Our primary outcomes of interest were the incidence of major bleeding events and time spent in therapeutic range (INR between 2–3) as calculated by the linear interpolation method,45 an accepted measure of anticoagulation quality and a potential surrogate for bleeding risk.9,11 Our secondary outcomes of interest included incidence of minor bleeding and thromboembolism, and measures of over-anticoagulation and inadequate anti-coagulation, including: time to first therapeutic INR, time to stable warfarin dose, percentage time INR greater than 3 or 4, and relative number of INR blood draws. We resolved all disagreements by discussion and consensus. We contacted the authors of the studies for additional information.
We used standard definitions for several outcomes. Major bleeding was categorized according to an international consensus statement46 as any of the following: fatal bleeding, symptomatic bleeding in a critical area or organ, a fall of hemoglobin greater than or equal to 2 mg/dl, or requirement of transfusion of two or more units of red cells or whole blood. For one study,47 we categorized gastrointestinal bleeds as major bleeds because we were unable to ascertain the severity of events reported. Percentage time in therapeutic range INR was defined between 2 and 3 unless otherwise stated. Time to stable warfarin dose was defined as two consecutive, therapeutic INR values separated by at least 7 days without intervening dose alteration.48
Study quality was assessed using the Jadad scale.49 The score ranged from 0 to 5 with higher scores indicating higher quality.Footnote 1 We further characterized study quality with assessment of: allocation concealment, comparability of baseline groups, equivalency of loss to follow-up, similarity of co-interventions and whether the analysis was intention to treat.
Data Synthesis and Statistical Analysis
Quantitative data synthesis was performed using STATA version 10 (STATA Corp, College Station, TX). Meta-analysis was performed on primary outcomes. The principal measures of effect size between the intervention and control arms were risk ratio for major bleeding, and the standardized mean difference (SMD) for percentage time INR within therapeutic range (online Appendix 2). In one case,48 we used a weighted average of the percentage time within therapeutic range from both the initiation (first 8 days) and stabilization period (day 9 through stable dose), because only separate estimates were reported in the study. We used a random effects model50 to combine results across studies when appropriate and assessed heterogeneity with the Q statistic51 and I2.52–54 In order to test for publication bias, we used the Begg adjusted rank correlation test and the Egger regression asymmetry test.55,56 Pre-specified subgroups for sensitivity analyses included the quality of the trials (poor versus good/excellent) and the length of follow-up (< 30 days versus ≥ 30). Sensitivity analysis by genes used in the prediction model was not performed because there were too few studies for it to be meaningful.
RESULTS
Study Characteristics
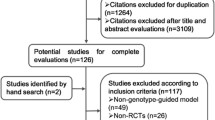
Our search identified 2,014 unique studies. Three studies satisfied all inclusion and no exclusion criteria (Fig. 1). All three studies were small, single-center randomized clinical trials ranging from 38 to 238 patients (Table 1). Follow-up ranged from 22 days in the pharmacogenetic arm of the study by Caraco et al. 48 to an average of 46 days across both arms of the study by Anderson et al.57 Patients were almost exclusively Caucasian (97%) older adults taking warfarin for the first time. Demographic characteristics were similar between studies (Table 2), but the treatment setting varied from predominantly outpatient to largely inpatient. Principal indications for warfarin initiation were atrial fibrillation, atrial flutter, deep venous thrombosis and pulmonary embolism in these three studies. Hillman et al.47 also included prosthetic valve and joint patients, and Anderson et al.57 included preoperative orthopedic patients. In all studies, observed genotype frequencies were in accordance with Hardy-Weinberg equilibrium.

Each of the three studies used different dosing models for their pharmacogenetic and control dosing arms, which is reflected in the differing average initial doses across the three studies in both groups (Table 1). For the pharmacogenetic arm, the studies by Hillman et al.47 and Caraco et al.48 used dosing models that accounted only for CYP2C9 variants, while Anderson et al.57 incorporated both CYP2C9 and VKORC1 variants. Two of the pharmacogenetic algorithms47–57 were previously validated and adjusted for covariates of age, sex and weight. In contrast, Caraco et al.48 created a new algorithm that estimated warfarin dose based only on CYP2C9 genotype and amiodarone use.
All three studies evaluated outcomes of bleeding and time within therapeutic range. No study reported active surveillance for clinical adverse events of bleeding or venous or arterial thromboembolism.
Study Quality
Study quality varied substantially (Table 3). Caraco et al.48 received the lowest Jadad score (1) for inadequate randomization and blinding. Patients in this study were randomized by the “even” or “odd” last digit of their identity number, and investigators were not blinded after day 8 of follow-up. The intention to treat principle was also violated: 51 excluded patients had initiated warfarin therapy, but were not included in the analyses, and there was no data comparing treatment groups at randomization. The authors followed the control arm, on average, for almost twice as long as the pharmacogenetic group (Table 1). However, time-dependent outcomes such as number of bleeding events, percent time in therapeutic range, time spent with out-of-range INR and total number of INR draws were not adjusted to account for different lengths of follow-up time.
The two remaining studies were of good quality overall. Hillman et al.47 received a Jadad score of 3 due to single-blinded design. Anderson et al.57 received the highest Jadad score (5). However, despite adequate randomization in the Anderson study,57 there was a significantly higher percentage of patients with ≥1 variant allele in CYP2C9 or VKORC1 among the control group compared to the pharmacogenetics group (p < 0.01, Table 2).
Primary Outcomes
Primary outcomes of interest were rates of major bleeding and percentage time INR in the therapeutic range (Table 4). None of the individual studies was powered to show a difference in major bleeding (Fig. 2), and the pooled risk ratio of 0.69 did not achieve statistical significance (95% CI 0.16 to 2.9). While there was no evidence of statistical heterogeneity (chi-squared p = 0.45, I2 = 0.0%), there was clinically important variability in the pharmacogenetic and control interventions, length of follow-up and study quality. Egger’s regression did not show evidence of publication bias (p = 0.79); however, this estimate is limited in the presence of only three studies (Online Appendix 3 shows estimates used for meta-analyses).

Forest plot. Meta-analysis of the risk ratio of major bleeding between pharmacogenetic dosing and the control group. This shows a pooled risk ratio of 0.69 favoring pharmacogenetics dosing, though it does not meet statistical significance (95% CI 0.16 to 2.9).
Figure 3 summarizes the differences in the percentage time in therapeutic range across the three studies. No pooled estimate is presented because there was significant heterogeneity (chi-squared p = 0.03, I2 = 72.5%). This outcome showed a strong beneficial effect in the Caraco study,48 but no effect in the other two.47,57 If the poor quality study is excluded, there is no longer significant heterogeneity (chi-squared p = 0.91, I2 = 0.0%), and the pooled estimate is not significant (p = 0.76). Notably, time within therapeutic range is as variable across the three studies as it is between intervention groups in the Caraco study48 (Table 4). Other sources of heterogeneity include differences in treatment algorithms and study quality, differing frequency of INR measurements and differing lengths of follow-up. Anderson et al.57 used a more lenient definition of therapeutic INR (1.8 to 3.2), which may also contribute to heterogeneity. Sensitivity analysis using follow-up time of 30 days and a standard definition of therapeutic INR (2 to 3) yielded a similar result (not shown). Heterogeneity remained borderline significant (p = 0.1, I2 = 57.4%), suggesting that the variability in study results was due to factors other than differences in follow-up time or INR.

Forest plot. Meta-analysis of average percentage time spent in the therapeutic range. The SMD is the difference in time spent in therapeutic range as a proportion of the standard deviation around the average value for the entire group. Here, no summary estimate is shown due to significant heterogeneity (chi-squared p = 0.03, I2 = 72.5%).
Secondary Outcomes
Overall, there were few consistent trends showing a difference in secondary outcomes between the groups. Caraco et al.48 reported an advantage for pharmacogenetic dosing compared to the control group for several outcomes (Table 4), including lower cumulative incidence of minor bleeding, decreased time to first therapeutic INR, decreased time to stable warfarin dose, decreased total number of INR draws and fewer days of INR >3 (1.77 vs. 6.58, p < 0.001). However, the longer follow-up time in the control arm complicates the interpretation of time-dependent results, including the number of bleeding events, total number of INR draws and days of supratherapeutic INR. It is noteworthy that the Hillman47 and Anderson57 studies did not replicate these findings. No study showed a difference in thromboembolism incidence.
The pharmacogenetic dosing groups showed improvement in time to stable warfarin dose compared to the control groups (Table 4) in two of the three studies48,57 and was not reported in the third.47 Among the studies reporting this outcome, the pharmacogenetic arm was favored with statistically significant results by Caraco et al.48 and near statistical significance by Anderson and colleagues (14.1 versus 19.6 days, p = 0.07).57 The longer follow-up in the control arm of the Caraco48 study did not affect this result.
Ongoing Clinical Trials
We identified at least five ongoing randomized clinical trials comparing pharmacogenetic dosing of warfarin therapy to a non-genetic control algorithm (Table 5). Notably, the National Heart, Lung and Blood Institute (NHLBI) is sponsoring a large (N = 1,238), multi-center, double-blinded, randomized trial comparing a recently validated38,58 clinical plus genotype-guided algorithm to a clinical only-guided dosing algorithm. In all, randomized control experience of pharmacogenetic dosing will encompass data collected from more than 2,500 patients.
DISCUSSION
Our study found little randomized trial data available to support the hypothesis that pharmacogenetic dosing at the onset of warfarin therapy reduces major bleeding events. An extensive search yielded only three small randomized trials evaluating pharmacogenetic dosing, and among these, there was significant variability in terms of design quality, length of follow-up, intervention and outcome measures. No study had adequate power to evaluate differences in major bleeding rates between groups. In the pooled estimates, there was a trend towards less bleeding with pharmacogenetic dosing, but this should be interpreted with caution because of the differences in design between studies. Percentage time within therapeutic range varied significantly across the studies even with standardized INR range and more uniform follow-up time. This disparity raises concern that methods of ascertainment of this outcome are likely to have differed between studies. There was some evidence that time to stable warfarin dose may be decreased with genotype-guided dosing.
The study by Anderson el al.57 is the highest quality trial published to date, and the only study that incorporated both VKORC1 and CYP2C9. It is notable that there were more variant alleles in the standard dosing arm compared to pharmacogenetic arm of this study.57 Because patients with variant alleles are known to be more likely to have out of range INR and bleeding complications, this difference could have biased the results in favor of the pharmacogenetic arm. Indeed, some outcome estimates in this trial favored pharmacogenetic dosing, but none achieved statistical significance.
Only the Caraco study48 showed statistically significant improvement in nearly all surrogate outcomes with pharmacogenetic dosing. However, the lack of true randomization and allocation concealment, the high loss to follow-up, the lack of intention to treat analysis and the different lengths of follow-up between groups challenge the internal validity of these results. Specifically, the outcomes of total number of bleeding events, percentage time INR in therapeutic range, days of supratherapeutic INR and total number of INR draws are invalidated on the basis of detection bias as a result of the nearly two-fold increased follow-up time for the control group. As an example, the total number of INR draws was 36% higher in the control group, but the average interval between consecutive INR draws was the same between groups.
Both the Hillman47 and Anderson57 studies used a multivariable algorithm to select the initial dose for the patients in the intervention arm, taking into account not only the contribution of genetic variation, but also other well-established factors that are known to affect overall warfarin dose such as age, sex and weight. In contrast, patients in the control arms of these two studies47,57 all received the same initial dose. Despite this seemingly unfair advantage at the outset, neither of these studies demonstrated statistically significant improvement of outcomes for the pharmacogenetic arm.
Is pharmacogenetic dosing of warfarin more safe and effective than a one-size-fits all strategy followed by careful INR monitoring? The results of our study demonstrate that we still do not know. An uncontrolled study 59 evaluating a CYP2C9 dosing algorithm 40 in patients initiating warfarin further highlights this uncertainty. Although the algorithm estimated the maintenance warfarin dose well (R2 = 0.42, p < 0.001), carriers of variant CYP2C9 alleles continued to have a significantly increased risk of INR >4 (HR 4.6, p < 0.01) compared to those with the wild-type allele. There is evidence that the greatest risk of warfarin-induced adverse events is at the induction of warfarin therapy,60 and that INR levels prior to day 4 of therapy do not predict dose response differences. Thus, the traditional “trial and error” method may result in delays in estimating the appropriate dose.41 However, Li and colleagues61 recently found that CYP2C9 and VKORC1 genotypes did not add to early INR response as a predictor of warfarin sensitivity. Even if pharmacogenetic dosing does not reduce major bleeding, it may still be useful and cost effective if it results in shorter time to stable dose, and fewer blood draws to attain stable INR. It is possible, however, that physicians may become more complacent with pharmacogenetic dosing resulting in reduced surveillance and a paradoxical increase in bleeding during the initiation of warfarin therapy.
Our study has limitations. First, very little high-quality evidence has been published in this area: we identified only three small randomized trials evaluating pharmacogenetic dosing of warfarin. Second, important differences in designs, outcome definitions and follow-up intervals used by these three trials reduced the degree to which we could pool their individual findings. We did not perform meta-analysis of secondary outcomes because of significant heterogeneity of the trials. Third, we did not evaluate genotype-specific outcomes, because these are not relevant when providers are unaware of genotypes in advance. Lastly, we did not include the three prospective cohort studies using genotype-guided algorithms,59,62,63 because others have reviewed this literature,64,65 and we decided a priori that only randomized trials could reliably demonstrate whether pharmacogenetic dosing improves patient outcomes. It may be considered early to perform a systematic review on a topic where so few randomized controlled trials are available; however, given the FDA relabeling, we feel it is important to evaluate the current evidence.
The package insert of warfarin advises that “lower initiation doses should be considered for patients with certain genetic variations in CYP2C9 and VKORC1 enzymes.” This FDA labeling change was made on the basis of accumulation of data66 demonstrating that allelic variants in CYP2C9 and VKORC1 are associated with increased plasma warfarin levels, out of range INR and increased bleeding risk.15,19,23,35,67,68 However, as our study demonstrates, there is no evidence that a more accurate initiation dose reduces the risk of bleeding. Results from ongoing clinical trials will help to clarify the role of genetic testing in warfarin management. A target enrollment of at least 2,000 patients has been suggested,57 and currently the cumulative experience of >2,500 patients is anticipated.
Each of the randomized trials reviewed in our study used different pharmacogenetic and control group dosing algorithms. The most comprehensive and widely available pharmacogenomic algorithm, http://www.WarfarinDosing.org, has been recently validated by the The International Warfarin Pharmacogenetics Consortium and will be used in the largest randomized trial sponsored by the NHLBI.38 Until recently, however, there was no widely accepted pharmacogenetic algorithm to guide the initiation of warfarin therapy, and new models are still being developed and validated.69 Although warfarin dosing algorithms do not eliminate the need for frequent INR monitoring and dose titration, these algorithms can, even in the absence of genotype information, provide a very good estimate of the patient’s warfarin dose by taking into account readily available information such as age, gender, weight and smoking status.38 Whether these algorithms improve outcomes compared to other warfarin initiation strategies is not known.
The products of genetic discovery are becoming increasingly relevant to the practice of clinical medicine, particularly in the realm of pharmacogenetics. Genotype-guided warfarin prescribing is currently the focus of much attention and is positioned to set a precedent for how integration of genetic technologies in clinical practice will proceed. In the case of warfarin, it seems intuitive that adjusting warfarin dose to match patients’ genetic makeup will result in fewer complications; however, our review, along with at least one unfavorable cost-effectiveness analysis,44 demonstrates that additional clinical trial data are needed prior to endorsing a new standard of care for warfarin dosing.
CONCLUSION
In conclusion, our study did not find sufficient evidence to support the use of pharmacogenetics to guide warfarin therapy outside of clinical trials at this time. Small sample sizes and heterogeneity across the few available studies precluded definitive estimates of the relative effectiveness of this intervention. We recommend that policy makers and clinicians await the results of larger, high quality randomized trials and better cost-effectiveness analyses before adopting genetic testing as the standard of care for warfarin initiation.
Notes
The Jadad score rates randomized controlled trials on a 0–5 scale where a higher score indicates higher quality and gives 1 point for each of the following: (1) the study was described as randomized, (2) the randomization technique was described and appropriate, (3) the study was described as double-blind, (4) the method of double-blinding was described and was appropriate, and (5) withdrawals and reasons for withdrawals were given.
References
FDA Approves Updated Warfarin (Coumadin) Prescribing Information: New Genetic Information May Help Providers Improve Initial Dosing Estimates of the Anticoagulant for Individual Patients. http://www.fda.gov/bbs/topics/NEWS/2007/NEW01684.html. Accessed February 19, 2009.
Verigene Warfarin Test. http://www.nanosphere.us/VerigeneWarfarinMetabolismNucleicAcidTest_4472.aspx. Accessed February 19, 2009.
Kimball Warfarin Test. http://www.kimballgenetics.com/pdf/WarfarinEdusheetv7.pdf. Accessed February 19, 2009.
Genelex Warfarin Test. http://www.healthanddna.com/drug-safety/warfarin.html. Accessed February 19, 2009.
McCabe LL, McCabe ER. Direct-to-consumer genetic testing: access and marketing. Genet Med. 2004;6(1):58–9.
Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med. 2007;167(13):1414–9.
Voora D, McLeod HL, Eby C, Gage BF. The pharmacogenetics of coumarin therapy. Pharmacogenomics. 2005;6(5):503–13.
Vecsler M, Loebstein R, Almog S, et al. Combined genetic profiles of components and regulators of the vitamin K-dependent gamma-carboxylation system affect individual sensitivity to warfarin. Thromb Haemost. 2006;95(2):205–11.
Ansell J, Hirsh J, Dalen J, et al. Managing oral anticoagulant therapy. Chest. 2001;119(1 Suppl):22S–38S.
Fihn SD, McDonell M, Martin D, et al. Risk factors for complications of chronic anticoagulation. A multicenter study. Warfarin Optimized Outpatient Follow-up Study Group. Ann Intern Med. 1993;118(7):511–20.
Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation. 2003;107(12):1692–711.
Landefeld CS, Beyth RJ. Anticoagulant-related bleeding: clinical epidemiology, prediction, and prevention. Am J Med. 1993;95(3):315–28.
Risk factors for stroke and efficacy of antithrombotic therapy in atrial fibrillation. Analysis of pooled data from five randomized controlled trials. Arch Intern Med. Jul 11 1994;154(13):1449–1457.
Bodin L, Verstuyft C, Tregouet DA, et al. Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxide reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity. Blood. 2005;106(1):135–40.
Sconce EA, Khan TI, Wynne HA, et al. The impact of CYP2C9 and VKORC1 genetic polymorphism and patient characteristics upon warfarin dose requirements: proposal for a new dosing regimen. Blood. 2005;106(7):2329–33.
Takahashi H, Wilkinson GR, Nutescu EA, et al. Different contributions of polymorphisms in VKORC1 and CYP2C9 to intra— and inter-population differences in maintenance dose of warfarin in Japanese, Caucasians and African-Americans. Pharmacogenet Genomics. 2006;16(2):101–10.
Hirsh J, Dalen J, Anderson DR, et al. Oral anticoagulants: mechanism of action, clinical effectiveness, and optimal therapeutic range. Chest. 2001;119(1 Suppl):8S–21S.
Takahashi H, Echizen H. Pharmacogenetics of warfarin elimination and its clinical implications. Clin Pharmacokinet. 2001;40(8):587–603.
Rieder MJ, Reiner AP, Gage BF, et al. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N Engl J Med. 2005;352(22):2285–2293.
Sanderson S, Emery J, Higgins J. CYP2C9 gene variants, drug dose, and bleeding risk in warfarin-treated patients: a HuGEnet systematic review and meta-analysis. Genet Med. 2005;7(2):97–104.
D’Andrea G, D’Ambrosio RL, Di Perna P, et al. A polymorphism in the VKORC1 gene is associated with an interindividual variability in the dose-anticoagulant effect of warfarin. Blood. 2005;105(2):645–9.
Veenstra DL, You JH, Rieder MJ, et al. Association of Vitamin K epoxide reductase complex 1 (VKORC1) variants with warfarin dose in a Hong Kong Chinese patient population. Pharmacogenet Genomics. 2005;15(10):687–91.
Wadelius M, Chen LY, Downes K, et al. Common VKORC1 and GGCX polymorphisms associated with warfarin dose. Pharmacogenomics J. 2005;5(4):262–70.
Yin T, Miyata T. Warfarin dose and the pharmacogenomics of CYP2C9 and VKORC1 - rationale and perspectives. Thromb Res. 2007;120(1):1–10.
Kaminsky LS, Zhang ZY. Human P450 metabolism of warfarin. Pharmacol Ther. 1997;73(1):67–74.
Lee CR, Goldstein JA, Pieper JA. Cytochrome P450 2C9 polymorphisms: A comprehensive review of the in-vitro and human data. Pharmacogenetics. 2002;12(3):251–63.
Tabrizi AR, Zehnbauer BA, Borecki IB, McGrath SD, Buchman TG, Freeman BD. The frequency and effects of cytochrome P450 (CYP) 2C9 polymorphisms in patients receiving warfarin. J Am Coll Surg. 2002;194(3):267–73.
Cain D, Hutson SM, Wallin R. Assembly of the warfarin-sensitive vitamin K 2,3-epoxide reductase enzyme complex in the endoplasmic reticulum membrane. J Biol Chem. 1997;272(46):29068–75.
Zhu Y, Shennan M, Reynolds KK, et al. Estimation of warfarin maintenance dose based on VKORC1 (−1639 G>A) and CYP2C9 genotypes. Clin Chem. 2007;53(7):1199–205.
Borgiani P, Ciccacci C, Forte V, Romano S, Federici G, Novelli G. Allelic variants in the CYP2C9 and VKORC1 loci and interindividual variability in the anticoagulant dose effect of warfarin in Italians. Pharmacogenomics. 2007;8(11):1545–50.
Aquilante CL, Langaee TY, Lopez LM, et al. Influence of coagulation factor, vitamin K epoxide reductase complex subunit 1, and cytochrome P450 2C9 gene polymorphisms on warfarin dose requirements. Clin Pharmacol Ther. 2006;79(4):291–302.
Schwarz UI, Ritchie MD, Bradford Y, et al. Genetic determinants of response to warfarin during initial anticoagulation. N Engl J Med. 2008;358(10):999–1008.
Ogg MS, Brennan P, Meade T, Humphries SE. CYP2C9*3 allelic variant and bleeding complications. Lancet. 1999;354(9184):1124.
Lindh JD, Lundgren S, Holm L, Alfredsson L, Rane A. Several-fold increase in risk of overanticoagulation by CYP2C9 mutations. Clin Pharmacol Ther. 2005;78(5):540–50.
Higashi MK, Veenstra DL, Kondo LM, et al. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA. 2002;287(13):1690–8.
Aithal GP, Day CP, Kesteven PJ, Daly AK. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet. 1999;353(9154):717–9.
Limdi NA, McGwin G, Goldstein JA, et al. Influence of CYP2C9 and VKORC1 1173C/T genotype on the risk of hemorrhagic complications in African-American and European-American patients on warfarin. Clin Pharmacol Ther. 2008;83(2):312–21.
Estimation of the Warfarin Dose with Clinical and Pharmacogenetic Data: The International Warfarin Pharmacogenetics Consortium. N Engl J Med. 2009 2009;360(8):753–64.
Caldwell MD, Berg RL, Zhang KQ, et al. Evaluation of genetic factors for warfarin dose prediction. Clin Med Res. 2007;5(1):8–16.
Gage BF, Eby C, Milligan PE, Banet GA, Duncan JR, McLeod HL. Use of pharmacogenetics and clinical factors to predict the maintenance dose of warfarin. Thromb Haemost. 2004;91(1):87–94.
Hamberg AK, Dahl ML, Barban M, et al. A PK-PD model for predicting the impact of age, CYP2C9, and VKORC1 genotype on individualization of warfarin therapy. Clin Pharmacol Ther. 2007;81(4):529–38.
Millican EA, Lenzini PA, Milligan PE, et al. Genetic-based dosing in orthopedic patients beginning warfarin therapy. Blood. 2007;110(5):1511–5.
Wadelius M, Chen LY, Lindh JD, et al. The largest prospective warfarin-treated cohort supports genetic forecasting. Blood. Jun 23 2008.
Eckman MH, Rosand J, Greenberg SM, Gage BF. Cost-effectiveness of using pharmacogenetic information in warfarin dosing for patients with nonvalvular atrial fibrillation. Ann Intern Med. 2009;150(2):73–83.
Rosendaal FR, Cannegieter SC, van der Meer FJ, Briet E. A method to determine the optimal intensity of oral anticoagulant therapy. Thromb Haemost. 1993;69(3):236–9.
Schulman S, Kearon C. Definition of major bleeding in clinical investigations of antihemostatic medicinal products in non-surgical patients. J Thromb Haemost. 2005;3(4):692–694.
Hillman MA, Wilke RA, Yale SH, et al. A prospective, randomized pilot trial of model-based warfarin dose initiation using CYP2C9 genotype and clinical data. Clin Med Res. 2005;3(3):137–45.
Caraco Y, Blotnick S, Muszkat M. CYP2C9 Genotype-guided Warfarin Prescribing Enhances the Efficacy and Safety of Anticoagulation: A Prospective Randomized Controlled Study. Clin Pharmacol Ther. Sep 12 2007.
Jadad AR, Moore RA, Carroll D, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary? Control Clin Trials. 1996;17(1):1–12.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–88.
Sutton AJ AK, Jones DR, Sheldon RA, Song F. Methods for Meta-analysis in Medical Research: Assessing Between Study Heterogeneity. West Sussex, England: John Wiley & Sons; 2000.
Higgins J, Thompson S, Deeks J, Altman D. Statistical heterogeneity in systematic reviews of clinical trials: a critical appraisal of guidelines and practice. J Health Serv Res Policy. 2002;7(1):51–61.
Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21(11):1539–58.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–60.
Begg CB, Mazumdar M. Operating characteristics of a rank correlation test for publication bias. Biometrics. 1994;50(4):1088–101.
Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ. 1997;315(7109):629–34.
Anderson JL, Horne BD, Stevens SM, et al. Randomized Trial of Genotype-Guided Versus Standard Warfarin Dosing in Patients Initiating Oral Anticoagulation. Circulation. Nov 7 2007.
Gage BF, Eby C, Johnson JA, et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin Pharmacol Ther. 2008;84(3):326–31.
Voora D, Eby C, Linder MW, et al. Prospective dosing of warfarin based on cytochrome P-450 2C9 genotype. Thromb Haemost. 2005;93(4):700–5.
Landefeld CS, Goldman L. Major bleeding in outpatients treated with warfarin: incidence and prediction by factors known at the start of outpatient therapy. Am J Med. 1989;87(2):144–52.
Li C, Schwarz UI, Ritchie MD, Roden DM, Stein CM, Kurnik D. Relative contribution of CYP2C9 and VKORC1 genotypes and early INR response to the prediction of warfarin sensitivity during initiation of therapy. Blood. Dec 12 2008.
Lenzini PA, Grice GR, Milligan PE, et al. Laboratory and clinical outcomes of pharmacogenetic vs. clinical protocols for warfarin initiation in orthopedic patients. J Thromb Haemost. 2008;6(10):1655–62.
Wen MS, Lee M, Chen JJ, et al. Prospective Study of Warfarin Dosage Requirements Based on CYP2C9 and VKORC1 Genotypes. Clin Pharmacol Ther. Jan 9 2008.
Lindh JD, Holm L, Andersson ML, Rane A. Influence of CYP2C9 genotype on warfarin dose requirements-a systematic review and meta-analysis. Eur J Clin Pharmacol. Nov 25 2008.
McClain MR, Palomaki GE, Piper M, Haddow JE. A rapid-ACCE review of CYP2C9 and VKORC1 alleles testing to inform warfarin dosing in adults at elevated risk for thrombotic events to avoid serious bleeding. Genet Med. 2008;10(2):89–98.
Lesko LJ. Regulatory Perspective on Warfarin Relabeling with Genetic Information. http://www.fda.gov/cder/genomics/presentations/Lesko200705.pdf. Accessed February 19, 2009.
Herman D, Locatelli I, Grabnar I, et al. Influence of CYP2C9 polymorphisms, demographic factors and concomitant drug therapy on warfarin metabolism and maintenance dose. Pharmacogenomics J. 2005;5(3):193–202.
Linder MW, Looney S, Adams Iii JE, et al. Warfarin dose adjustments based on CYP2C9 genetic polymorphisms. J Thromb Thrombolysis. 2002;14(3):227–32.
CReating an Optimal Warfarin Nomogram (CROWN) Trial. NCT00401414. http://www.clinicaltrials.gov/ct2/show/NCT00401414?term = %28warfarin + OR + coumadin%29 + AND + %28genotype + OR + gene + OR + pharmacogenetic%29&rank = 9. Accessed February 19, 2009.
Hillman MA, Wilke RA, Caldwell MD, Berg RL, Glurich I, Burmester JK. Relative impact of covariates in prescribing warfarin according to CYP2C9 genotype. Pharmacogenetics. 2004;14(8):539–47.
Assessing impact of organizational interventions — Marshfield Clinic’s Coumadin Clinic Evaluation: Final Report.: Marshfield Medical Research and Education Foundation; 13, May 2001.
Ageno W, Johnson J, Nowacki B, Turpie AG. A computer generated induction system for hospitalized patients starting on oral anticoagulant therapy. Thromb Haemost. 2000;83(6):849–852.
Carlquist JF, Horne BD, Muhlestein JB, et al. Genotypes of the cytochrome p450 isoform, CYP2C9, and the vitamin K epoxide reductase complex subunit 1 conjointly determine stable warfarin dose: a prospective study. J Thromb Thrombolysis. 2006;22(3):191–7.
Kovacs MJ, Rodger M, Anderson DR, et al. Comparison of 10-mg and 5-mg warfarin initiation nomograms together with low-molecular-weight heparin for outpatient treatment of acute venous thromboembolism. A randomized, double-blind, controlled trial. Ann Intern Med. 2003;138(9):714–9.
Acknowledgements
The authors are indebted to Gloria Won from the Fishbon Memorial Library for her expertise and efforts in creating a highly sensitive search strategy. We express our appreciation to Benjamin Horne and Jeffery Anderson for providing us additional data from their study. At the time the review was conducted Dr. Kangelaris was supported by the National Research Service Award Institutional Grant (T32 HP19025).
Conflict of Interest
None disclosed.
Author information
Authors and Affiliations
Corresponding author
Additional information
None of the authors has any conflicts of interest to disclose. At the time the review was conducted, Dr. Kangelaris was supported by the National Research Service Award Institutional Grant (T32 HP19025). Findings were presented at the Society of General Internal Medicine meeting on April 10, 2008.
An erratum to this article can be found at http://dx.doi.org/10.1007/s11606-009-1081-y
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 69.0 KB)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kangelaris, K.N., Bent, S., Nussbaum, R.L. et al. Genetic Testing Before Anticoagulation? A Systematic Review of Pharmacogenetic Dosing of Warfarin. J GEN INTERN MED 24, 656–664 (2009). https://doi.org/10.1007/s11606-009-0949-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11606-009-0949-1