
Abstract
Introduction
Hepatic adenomas (HAs) are benign tumors of the liver, which can be solitary or multiple, and have a definite risk of malignant degeneration.
Discussion
The pathogenesis and natural history of this disease entity were previously unknown. Recent research into the molecular pathogenesis of this condition has provided evidence for the malignant transformation of some of these adenomas. In the current article, we discuss the current evidence on the molecular biology underlying malignant transformation of hepatic adenomas and the implications for the surgical management of this disease.
Similar content being viewed by others


Introduction
Hepatic adenoma (HA) is a benign neoplasm usually occurring in an otherwise normal liver. There are two entities described so far. The first condition is solitary adenoma associated with the use of oral contraceptives or anabolic steroids, and the second is hepatic adenomatosis, first described by Flejou and colleagues, whereby the patient develops multiple adenomas (usually ten or more).1, 2 Uncomplicated adenomas are usually asymptomatic making their true prevalence difficult to assess. The two main complications of hepatic adenoma are hemorrhage from rupture and malignant transformation. The current treatment strategy for hepatic adenomas is poorly defined. Traditionally, the treatment for large, solitary hepatic adenomas will be surgical resection.3 However, in patients who have asymptomatic, small lesion or multifocal disease not amenable to resection, the treatment strategy is less clear-cut. The controversy that plagues the “watch-and-see” approach is the risk of malignant transformation, which is hitherto poorly characterized. The purpose of the current review is to summarize our current understanding of the molecular biology of hepatic adenomas in an attempt to address the fundamental question if hepatic adenomas are premalignant and can transform into a cancer. This review will provide the background for future rationalization of the treatment of this disease entity.
Methods
A literature search was performed using PubMed (1960 to 2013) with the keywords “hepatic adenomas,” “liver cell adenomas,” “pathogenesis,” “etiology,” “malignant transformation,” “treatment,” and/or “surgical treatment.” We focused on articles addressing the pathogenesis of hepatic adenomas and the process of malignant transformation. Articles discussing the etiology and pathogenesis of hepatocellular carcinoma were excluded. Further, studies reporting case/s of hepatic adenoma with malignant transformation were identified. The references of identified articles were assessed for further relevant papers. In Table 1, the clinical cases found to have malignant transformation of hepatic adenomas were summarized, with level of evidence provided according to the Oxford Centre for Evidence-Based Medicine when applicable.4
Epidemiology and Risk Factors for Hepatic Adenomas
HAs are rare tumors which are more common in females, with an estimated incidence of approximately 1 case per 100,000 women.5 In the past, the incidence of HAs rose following the introduction of oral contraceptives. It was estimated that the annual incidence of HA was 3–4 women per 100,000 users of oral contraceptives.6 However, the potency of oral contraceptives has since been reduced, and accounts for a reduction in the incidence of HA. Female gender, oral contraceptives, and anabolic steroids are risk factors for development of HA. Interestingly, the development of hepatic adenomatosis (multiple, >10 adenomas), which was previously thought to be a separate clinical entity, has little correlation with steroid use and has equal incidence in male and female gender.1 Glycogen storage diseases, especially Type I (von Gierke) and Type III, are well-known risk factors for hepatic adenomatosis development.7
Association with Estrogen Therapy
Numerous previous reports have associated young women on oral contraceptives with a tendency to develop HAs. More importantly, withdrawal of estrogen therapy has been associated with regression of the HA, even if when they are of considerable size.8–11 The roles of estrogen in the development of hepatic adenomas were speculated on the basis of several clinical observations. Firstly, history of hormone therapy was confirmed between 55 and 77 % of patients with hepatic adenomas. Secondly, there is a relatively rare incidence of hepatic adenoma in male patients. Together, this implicated the potential roles of estrogen and its cognate receptors in stimulating hepatocyte proliferation and HA formation. Further, estrogen and progesterone receptors have been found in HA lesions.12 However, there is variable expression of estrogen receptors (26–73 %) in hepatic adenomas. As such, it is not surprising some hepatic adenomas do not regress even with withdrawal of hormone therapy.
Association with Glycogen Storage Disease
Glycogen storage diseases (GSD) are inherited autosomal recessive disorders characterized by intracellular accumulation of glycogen. Type I GSD (von Gierke’s disease) is the most common form, and results from defects in glucose-6-phosphatase system. Type III GSD on the other hand is due to deficiency of glycogen-debranching enzyme. Both diseases result in impaired glycogenolysis and excessive glycogen accumulation in organs, especially in the liver and kidney. Approximately 22–75 % of patients with Type I GSD develop hepatic adenomas, usually diagnosed by the second decade of life.7 In Type III GSD, only 4.4–25 % of the patients develop HAs.13, 14 Further, the estimated risk of malignant transformation of Type I GSD-related HAs is as high as 10 %.7 The risk of HCC development is very low in Type III GSD as to date, there are only six cases of HCC reported in the literature.13 The biological consequence of excessive glycogen storage seems to induce carcinogenesis in liver. This is supported by animal studies using a rat model by Bannasch and colleagues who showed the concept that glycogenosis is an early event in liver cells during hepatic carcinogenesis induced by N-nitrosomorpholine.15, 16 This is accompanied by distinct changes in expression of carbohydrate metabolizing enzymes in rat liver. As such, hepatic glycogenosis may be causally related with hepatic adenoma-carcinoma progression in Type I GSD patients, but the exact mechanism is uncertain.
Pathology of Hepatic Adenomas
Classically, hepatic adenoma is characterized by histological appearance of monotonous sheets of hepatocytes, in the absence of biliary components, fibrosis, or dysplasia. It has been described that hepatic adenomas are consisted of monoclonal cells, whilst focal nodular hyperplasia consists of polyclonal cells.17 In general, HAs which develop in patients following exogenous hormonal therapy (e.g., oral contraceptives) are large, single, and encapsulated, whilst those associated with GSD are small, multiple, and unencapsulated.18 It is important to note that well-differentiated hepatocellular carcinoma can mimic the appearance of hepatic adenoma.
Adenoma-Carcinoma Sequence of Hepatic Adenomas
There have been multiple reports of malignant change in HAs. Detailed pathological analysis of sections of the adenomas with malignant transformation has revealed that the cancerous lesions occur within regions of otherwise typical adenoma.19 As such, these cancerous changes are not coincidental occurrence of synchronous lesions within the liver. To support this, recent molecular analysis of the hepatic adenomas revealed the same nucleotide mutation of β-catenin in the adenoma and HCC part the adenoma, indicating that the malignant transformation evolved from the original benign adenoma and suggesting the same clonal origin for the lesions.20 This conclusion has recently been replicated by Michelli and colleagues who found that hepatocellular carcinomas were found directly within hepatic adenomas, implicating a process of degeneration into malignancy.21
At least in cases of oral contraceptive-associated hepatic adenoma, the prolonged use of contraceptive may lead to dysplastic changes within hepatic adenomas that may eventually progress into hepatocellular carcinoma.19, 22 The key finding that supports the adenoma-carcinoma sequence in hepatic adenoma is the discovery of foci of dysplasia within adenomas.19 Although this remains controversial, it is thought that the development of dysplasia within a hepatic adenoma commits the adenoma towards an irreversible process of malignant degeneration. As such, the development of dysplastic changes within a hepatic adenoma can be likened to the phenomenon of carcinoma-in-situ in the natural history of other epithelial cancers.23, 24 Although the development of liver cell dysplasia is commonly associated with cirrhotic livers, it can develop in other liver conditions, such as HAs. This hypothesis explains the cases whereby the hepatic adenomas regress initially following withdrawal of hormonal therapy; however, HCC develops subsequently. A plausible explanation is that hepatic adenoma is a disease entity that is reversible, which accounts for the shrinkage of adenomas following withdrawal of growth stimulus such as hormonal therapy. If however there is development of dysplasia within the adenoma, this entity will persist and eventually develops into a cancer, accounting for the malignant recurrence at the site of hepatic adenomas.
In hepatic adenomatosis, there has been description of malignant degeneration as well. In 2000, Chiche and colleagues reported on their eight cases of hepatic adenomatosis and reviewed all previous cases of adenomatosis.25 Given the small series, there was a single case of malignant transformation within this group. In cases of Type I GSD, HCC develops in the context of hepatic adenomatosis, and the mechanism of this is believed to be adenoma-carcinoma progression.26, 27 On the other hand, Type III GSD is associated with liver cirrhosis and HCC develops against this background of cirrhosis (cirrhosis-carcinoma sequence), rather than from adenomas13 (Fig. 1).

Two routes of malignant transformation within the liver. Apart from the cirrhosis-associated hepatocellular carcinoma development, it is now thought that there is the existence of the adenoma-carcinoma sequence, likened to the phenomenon of carcinoma-in-situ in the natural history of other epithelial cancers. It is plausible that hepatic adenoma is a disease entity that is reversible, which accounts for the shrinkage of adenomas following withdrawal of growth stimulus such as hormonal therapy. However, the development of dysplasia within a hepatic adenoma commits the adenoma towards an irreversible process of malignant degeneration
Cells of Origin of Hepatic Adenomas
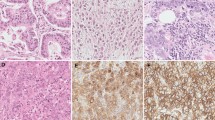
Recently, there has been a paradigm shift in that cancer is increasingly thought to arise from a subgroup of cells known as the “cancer stem cells.” The cancer stem cell theory states that the transformed stem cells retain their renewal capacity and continuously proliferate and differentiate to recapitulate the tumor.28 It is plausible that hepatic adenomas can arise from hepatic stem cells or committed progenitor cells. Whether hepatic adenomas and hepatocellular carcinomas develop from the same stem cells remain to be elucidated. Hepatic progenitor cells may have a role in development of hepatic tumors. Interestingly, it was found that hepatic progenitor cells and intermediate hepatocyte-like cells were discovered in 50 % of dysplastic hepatic foci and hepatic adenomas.29 This suggests that at least in some instances, HAs can arise from hepatic progenitor cells, which when transformed can give rise to dysplastic foci and ultimately, hepatocellular carcinomas.30 This further implies that HA and HCC can have a common cell of origin and that some of the HAs may be precursor lesions for HCC (Figs. 2 and 3).

β-catenin activation in hepatic adenomas predisposes to malignant degeneration. a In normal hepatocytes, the levels of β-catenin are controlled by its degradation following phosphorylation by GSK-3β of its “degradation domain” (serine/threonine phosphorylation site). b Mutations in the CTNNB1gene at the “degradation domains” lead to protein stabilization, and the mutant β-catenin resists degradation leading to its nuclear accumulation. Nuclear accumulation will lead to persistent activation of the β-catenin pathway which results in autonomous growth of hepatocytes

Surgical management of hepatic adenoma based on currently available clinical and molecular risk factors for malignant transformation
Risk of Malignant Transformation of Hepatic Adenomas
The risk of malignant transformation of hepatic adenomas is largely unknown. In a multicentre analysis of five academic hepato-pancreato-biliary units in North America, 5 out of a series of 124 patients (over a 10-year study epoch) with hepatic adenoma had underlying malignancy proven pathologically following liver resection.31 Of note, in cases of hepatic adenomas whereby the primary lesion has regressed following withdrawal of hormone therapy, there is still a risk of malignant progression, given that there has been cases of late development of hepatocellular carcinomas several years after adenoma regression. The overall frequency of malignant transformation of HA has recently been estimated to be 4.2 % in a systematic review including 1,600 cases of HA.32 The high-risk categories include patients with GSD Type I, ongoing use of steroid, and male gender.33 The characteristics of described cases of HA with malignant progression are provided in Table 1 and Supplemental Online Table.
Genotype–Phenotype Classification of Hepatic Adenomas
The French collaborative network has devised a system of classifying hepatic adenomas based on a combination of genetic aberrations and histological appearance.20 They showed a strong genotype–phenotype correlation in a series of 96 analyzed cases. Hepatic adenomas can be classified into four main categories: (1) HNF1α inactivation, with phenotypic features of marked steatosis, lack of cytological atypia, and absence of inflammatory infiltrates (40–45 %), (2) β-catenin activation, with atypical features such as pseudoglandular formation (15–19 %), (3) inflammatory group with presence of acute inflammatory infiltrate (30–35 %), and (4) non-mutated HNF1α and β-catenin (unclassified) group without inflammatory infiltrate. Of note, Zucman-Rossi and colleagues have observed that no case of HAs appear to have both mutations in HNF1α and activation of β-catenin, suggesting that the two pathways may be mutually exclusive. More importantly, it was found that lesions with β-catenin activation have a higher risk of malignant transformation, compared to other subtypes.20 Inflammatory subtype of HA is predominantly found in female patients and frequently associated with alcohol use and obesity.20, 34 Importantly, a subgroup of inflammatory HAs can harbor β-catenin mutation and consequently have an increased malignant risk.35
From a biological standpoint, impaired fatty acid shuttling and metabolism lead to the histological phenomenon of steatosis associated with HAs.36 Mechanistically, the HNF1α gene controls fatty acid trafficking through its effect on the L-FABP (liver fatty acid binding protein). Hence, inactivation of HNF1α will result in impaired fatty acid transport, fat accumulation, and increased lipogenesis, contributing to the steatotic phenotype seen in HNF1α-inactivated HAs.36 Given that HAs with HNF1α inactivation are at a low risk of malignant transformation, Van der Borght and colleagues have suggested the use of histological steatosis as a surrogate marker for HNF1α inactivation and hence, identifying a subgroup of HAs with a lower risk of malignant transformation.37
Recently, Bioulac-Sage and colleagues showed the feasibility of using a number of immunohistochemical markers to help classify HAs according to the described classification system, with potentials for routine clinical practice.34 For routine clinical use, the detection of genetic mutations will need to be easily performed and the most common method widely available in pathology laboratory will be standard immunohistochemistry. Bioulac et al. developed several surrogate immunohistochemical markers to help classify the HAs. Absent L-FABP (a downstream molecular target of HNF1α) staining on tumor slides was used to identify HAs with HNF1α inactivation and β-catenin activation is indicated by nuclear accumulation of β-catenin staining in HAs.
More recently, Laumonier and colleagues attempted to use magnetic resonance imaging as a non-invasive means of identifying subtypes of HAs. They found that it was possible to identify HNF1α-mutated and inflammatory HAs as these lesions demonstrated characteristic features on MRI imaging. This was clearly useful as these two subtypes form 80 % of all HAs. As such, this adds a further dimension to our ability to classify or subtype HAs, which now consists of clinical, genotype, histological/immunohistochemical, and radiological features (Table 2). The characterization of subtypes of HAs based on the immunohistochemical profile of these genetic aberrations has been reproduced by several groups worldwide.35
This genotype–phenotype classification may help in identifying high-risk patients who may benefit from aggressive therapy, whilst HAs with low risk of malignant transformation may be conservatively managed. However, given that much of the current data are based on retrospective analysis, a prospective validation study should be conducted to assess the utility and safety of the genotype–phenotype classification system to allow development of clinical guidelines. The above work has set the stage for further elucidation of genetic aberrations in HAs, which we will discuss in subsequent sections.
Genetic Aberrations in Hepatic Adenomas
From the clinical perspective, the solitary HAs were once thought to be a different disease entity from hepatic adenomatosis (multiple HAs). However, it is now increasingly clear that the genetic basis of both conditions does not differ and their genetic mutation spectrum is largely similar.20 There is convincing evidence to suggest that HA is a genetic disease, and in particular, there is a genetic basis to its risk of malignant degeneration.
Chromosomal Aberrations in Hepatic Adenomas
Cytogenetic analysis of HAs using fluorescence in situ hybridization and comparative genomic hybridization has found occasional gains on 7p, 17q, 20p, and 20q and rarely, deletion of 8p.38, 39 In comparison to hepatocellular carcinoma, HAs have lesser degree of chromosomal aberrations, which supports the hypothesis that the spectrum from HA to malignancy involves a cumulative genetic aberrations. 39, 40 More specifically, loss of heterozygosity (LOH) for mannose 6-phosphate/insulin-like growth factor II receptor (M6P/IGFRII) has been found in both HA and HCC.41 Competent M6P/IGFRII signaling is required for the activation of transforming growth factor β (a tumor suppressor gene). Similarly, LOH in DNA mismatch repair gene, human MutL homologue-1 (hMLH1) was also identified in both HA and HCC.42 These findings suggest that allelic loss in HA may be an early event in the progression to HCC.
TCF1/HNF1α
The genetic basis of hepatic adenomas comes from the seminal work by Bluteau et al. that the most common genetic aberration seen in hepatic adenoma is a mutation that inactivates the TCF1 gene.43 The TCF1 gene encodes for the hepatocyte nuclear factor 1α (HNF1α). Biallelic inactivation of TCF1 gene is found in 35–50 % of hepatic adenomas.20, 34, 43 In these cases, the majority of them (84 %) has somatic mutations in both alleles, and the remaining cases have one of the TCF1 mutations being a germline mutation.44 The families with heterozygous germ-line inactivation of TCF1 gene will display the adenomatosis phenotype, in which individuals develop more that ten adenomas when they lose the heterozygosity for TCF1 gene.45 This observation is of crucial importance in that it describes the underlying genetic basis of hepatic adenomatosis, which may conform to the classic Knudson’s two-hit hypothesis. In short, it is probable that patients with germline mutation of TCF1 are predisposed to development of hepatic adenomatosis, given that the hepatocytes would only require one further mutation (i.e., “second hit”) to form an adenoma in the liver. This permissive condition probably led to the formation of multiple adenomas (hepatic adenomatosis). More importantly, this observation suggests that individuals with hepatic adenomatosis probably have a germline mutation in a gene that predispose to hepatic adenomas. However, not all kindred with TCF1 mutation will develop adenomatosis, indicating that germ line TCF1 mutation has an incomplete penetrance.45, 46 In short, the TCF1 gene is an early tumor suppressor gene that becomes inactivated during the development of hepatic adenoma. Of note, HAs with HNF1A mutations tend to be indolent and do not progress.
Cosegregation of diabetes and hepatic adenomas in a large family was previously described.47 More recently, heterozygous germline mutations in the TCF1 gene have been found to be present in patients with the maturity-onset diabetes of the young type 3 (MODY3).48 In the study by Bluteau et al., one of the patients in the study series have a germline mutation in TCF1 gene.43 This individual had clinical features suggestive of MODY3, and more interestingly, the patient had a family history of both diabetes and hepatic adenomatosis. The previously described association between hepatic adenomas and diabetes, specifically MODY3, can now be attributed to germline mutations in TCF1 gene, providing a genetic explanation for this association.
β-catenin/Wnt Signaling Pathway
The CTNNB1 gene encodes for β-catenin which is a key molecule in the Wnt signaling pathway. When β-catenin translocates into the nucleus, it can activate the transcription of a range of genes involved in hepatocyte physiology including cell proliferation, stem cell renewal, lineage specification, epithelial–mesenchymal transition, and cell adhesion.49, 50 In normal hepatocytes, the levels of β-catenin are controlled by its degradation following phosphorylation by GSK-3β of its “degradation domain” (serine/threonine phosphorylation site). However, mutations in the CTNNB1gene at the “degradation domains” lead to protein stabilization, and the mutant β-catenin resists degradation leading to its nuclear accumulation. Nuclear accumulation will lead to persistent activation of the β-catenin pathway which results in autonomous growth of hepatocytes. Mutations in β-catenin gene are present in 15–30 % of HAs.20, 51 Torbenson et al. found evidence of nuclear accumulation of β-catenin in some HAs, indicating activation of the Wnt signaling pathway.12 Importantly, HAs with β-catenin mutations seem to have a tendency to progress to HCC. This corroborates with the findings that β-catenin activation is a frequent (20–34 % of cases) event in HCCs.52–55 As such, the CTNNB1 gene may represent an oncogene in the natural history of progression from HA to HCC. HAs with β-catenin mutation are more likely to progress to HCC (up to 50 %), making β-catenin activation as a surrogate high-risk marker for HCC progression.20
Interleukin-6–Gp130 Signaling Pathway
Inflammatory features have been previously observed in a subgroup of HAs.20 This prompted Rebouissou et al. to screen HAs by genome-wide transcriptome analysis to identify inflammatory genes that may have role in the pathogenesis of this subtype of HAs.56 In a group of inflammatory HAs, activating mutations were found in interleukin-6 signal transducer (IL6ST) gene which encodes for the cell surface co-receptor, gp130 protein. Under normal physiological conditions, interleukin-6 (IL-6) binds to its cognate IL-6 receptor (IL-6R) which forms a hexameric complex consisting of wo molecules each of IL-6, IL-6R, and gp130. This hexameric complex will activate STAT3 transcriptional factor and its downstream targets. These IL6ST mutations found in inflammatory HAs resulted in constitutive activation of gp130 proteins which activate STAT3 and inflammatory response genes downstream, even in the absence of IL6. Interestingly, it was found that in a small group of HCCs with inflammatory features, there was evidence that gp130 mutations are accompanied by β-catenin-activating mutations, implying a possible cooperative effect of these signaling pathways in the malignant transformation of hepatocytes.56
APC/Familial Polyposis Coli
The APC protein functions as a component of the degradation complex together with Axin and GSK-3β to control the levels of β-catenin in the cell. If there is biallelic loss/mutation of APC gene, this will prevent degradation of β-catenin and hence, allow nuclear accumulation of β-catenin and its downstream activation. Three cases of HA have been previously described in FAP patients.5, 57, 58 In two of the cases, biallelic inactivation of APC gene leading to the activation of β-catenin was thought to be the driving mechanism for HA formation.57, 58 In the remaining case, the β-catenin pathway was not activated as there was only monoallelic inactivation of APC in that patient. However, it was found that the HA from this patient displayed biallelic inactivation of the HNF1α gene. Hence, both the APC/β-catenin/Wnt and HNF1α signaling pathways can promote HA formation in FAP patients.
p16INK4a and p14ARF Alterations
The INK4a-ARF (CDKN2A) locus on chromosome 9p21 encodes for two tumor suppressor proteins, p16INK4a and p14ARF, through distinct first exons and alternative reading frames of shared downstream exons. They are often inactivated in human cancers, including HCC. p16INK4a inhibits CDK4/6-mediated phosphorylation of retinoblastoma protein, thereby blocking entry into the S-phase of cell cycle. Given that p14ARF stabilizes p53 by inhibiting its MDM2-mediated proteasomal degradation, loss of p14ARF will lead to degradation of tumor suppressor p53 and hence, promotes tumorigenesis. Tannapfel and colleagues found that HAs showed evidence of epigenetic silencing through hypermethylation of p16INK4a and p14ARF gene promoters, in 12 and 24 % of the cases, respectively.59 Inactivation of these genes will lead to induction of cell cycle progression and proliferation in HAs, contributing a potential malignant degeneration.
Sex Steroid Receptors and Sex Steroid Metabolism Pathway
The development of HAs has been closely linked to past or current exposure to hormonal therapy. Jeannot et al. studied the genes involved in estrogen metabolism in hepatic adenomas and found that CYP1B1 germline heterozygous mutations are found in women with HNF1α-mutated adenomas.44 It is likely that the inactivating mutations in CYP1B1 can act synergistically with TCF1 mutations to increase the incidence of HAs. The CYP1B1 protein is responsible for hormone metabolism, and the inactivation of this protein through mutation can result in accumulation of estrogen metabolite that may have genotoxic effects, though the exact mechanism that predisposes to HA formation remains unknown.
MicroRNAs
MicroRNAs (miRNA) are short, endogenous, non-coding RNAs which can post-transcriptionally regulate expression of specific genes. The primary modes of action are by promoting degradation of specific mRNAs and hence, suppressing gene expression, as well as binding to 3′-untranslated region of the targeted mRNA to prevent translation. Ladiero et al. recently profiled the miRNA expression of hepatic adenomas.60 They found that HAs upregulate miR-224 in a similar fashion as HCCs. Furthermore, miR-122a was found to be downregulated in both HAs and HCCs. More importantly, they demonstrated using a cell line model that functionally HNF1α is capable of directly regulating the miR-107 expression. Further, based on their profiling data, they speculated that β-catenin activation can downregulate the miR-375 levels in HAs. Further work will need to be performed to clarify the roles of microRNAs in the pathogenesis of hepatic adenomas.
Experimental Models of Hepatic Adenomas
The MET gene encodes the receptor tyrosine kinase, Met. In an elegant study by Tward and colleagues, they showed that transgenic mice overexpressing the MET proto-oncogene developed both HA and HCC.61 However, it was observed that HCC did not develop within HAs. Using hydrodynamic transfection of transgenes, Tward and colleagues found that MET overexpression when combined with HNF1α inactivation leads to HAs in 50 % of the mice. On the other hand, overexpression of MET supplemented with β-catenin activation led to these mice developing HCC in 74 % of surviving mice. More interestingly, if the MET+/β-catenin + mice subsequently have MET downregulated, β-catenin activation alone is able to result in recurrent HCC. These experiments suggest that with the background of MET overexpression, HNF1α inactivation or β-catenin activation can induce HA or HCC, respectively. It is important to note that in the context of the MET transgenic mice, the genetic background is one that is artificial and constricted compared to the genetic diversity found in human HAs and indeed, HCC. Corroborating evidence from other groups showed that HNF1α knockout mice develops liver enlargement and hepatic dysfunction,62, 63 whilst in rat models of carcinogenesis, there is a decrease in HNF1α expression in preneoplastic liver nodules, implying the tumor suppressor roles of HNF1α in cellular growth of liver nodules.64
Management of Hepatic Adenomas
We have summarized the evidence, from both clinical and biological studies, that HAs are due to genetic aberrations and a subset of HAs are precursor lesions for HCC. However, it appears that HAs can take varying durations of time to progress to malignancy, and in some cases, progression can be very slow or rapid. The onus is now on the research that aims to risk-stratify these lesions into groups with low and high risk of malignant degeneration. This will facilitate development of strategies to follow-up and treat patients with HA. Zucman-Rossi and colleagues have recently suggested a clinical algorithm for the diagnosis and treatment of HAs, based on the genotype–phenotype classification system.35 Of note, this algorithm has not been validated prospectively, in particular with regard to sensitivity and specificity for detecting malignant change within HAs. In the absence of this evidence, the optimal management of HAs remains controversial. Any decision regarding the selection of conservative versus surgical approach will depend on the clinical context. However, the molecular analyses of HAs have contributed significantly by providing several molecular characteristics that predict high risk of malignant change. Corroborating the previously known clinical features and the new molecular data, surgical resection of HA is indicated if:
-
1.
Large size of HA (>5 cm) with the impending risk of rupture or hemorrhage
-
2.
Evidence of β-catenin activation in any HA
-
3.
Any HA in male patients
(The above three features are high risk factors for malignant change in HAs)
-
4.
Evidence of dysplasia or atypia within an adenoma.
-
5.
Clinical features of malignant transformation in HA (i.e., increasing size, malignant features on imaging)
Of note, the α-fetoprotein levels are often in the normal range (see Table 1). As such, this is a poor indicator of malignant transformation.
In cases of solitary HA with the above high-risk factors whereby resection can be safely performed, surgery should be offered as it provides a chance of long-term cure. The rationale for this is that resection prevents future occurrence of malignancy, as there has not been any report of HCC recurrence following resection of adenoma. Also, HAs, if unresected, will have a risk of malignant degeneration with time, even in cases where the adenoma has regressed. Lastly, resection also eliminates the risk of observing a lesion that has been incorrectly diagnosed as benign and will prevent future complications such as rupture and hemorrhage.
The management of hepatic adenomatosis (multiple HAs) will require a tailored approach given the lack of robust clinical data. From the genetic perspective, it is likely that hepatic adenomatosis have the same genetic mutation underpinning their development as solitary HAs. As such, it has been suggested that liver resection should be considered along the same principles as solitary of HAs, i.e., if the lesions are >5 cm in size or harbors β-catenin activation.35 Of note, there is a higher frequency of malignant transformation (and β-catenin activation) in hepatic adenomatosis arising in the context of GSD. Liver transplantation is an option for patients with GSD when the presence of progression to HCC in HA is confirmed, but with no evidence of metastatic disease.65
Given that biopsy specimen can be inconclusive and misleading as some lesions of HCC can be surrounded by HA tissue, and the differentiation of HA from well-differentiated HCC remains difficult on histopathological analysis, close follow-up of patients who are treated conservatively should be mandatory. Regular liver imaging with monitor of alpha-fetoprotein is essential, although alpha-fetoprotein level is only helpful if it is raised or rising. Although there are limited data on its use in HA cases, it is likely that there is a role for radiofrequency ablation in treatment of hepatic adenomas not amenable to resection. Further, hepatic arterial chemoembolization has been reported as effective sole therapy in treating multiple hepatic adenomatosis.66 or in cases of HAs which are not resectable (i.e., central lesions).67
Future Work
The future of the management of HAs will be a tailored patient management strategy based on the genotype–phenotype classification of the lesion. Although the genotype–phenotype classification of HAs represent a significant advance, the clinical applicability of this will need further validation and clarification, perhaps with a large multicentre prospective study. Given that most of the reported case series are surgical series whereby patients have undergone surgical therapy, there is an inherent bias towards patients who have larger adenomas, complicated or symptomatic disease, which prompted surgical attention. As such, our understanding of the natural history of this disease will be skewed towards the extreme spectrum of the disease. To solve this, perhaps a registry for hepatic adenoma patients should be set up, which includes any patient with the diagnosis whether treated or just being monitored. Long-term cohort study of such patients taking into account the genetic subtype of the HA will allow elucidation of the natural history of these lesions. This will clearly require a multicentre collaboration given the rarity of HAs. It is possible that further work will reveal increasing diversity and complexity to the genetic and biological aspects of the malignant transformation of hepatic adenomas. From the therapeutic standpoint, given that β-catenin is implicated in HAs with higher risk of malignant degeneration, perhaps development of β-catenin inhibitors will be a useful adjunct to our treatment strategy. For instance, β-catenin activated HAs can be treated with such inhibitor, as a prophylactic measure to prevent malignant transformation.68
Conclusions
Hepatic adenoma is probably a heterogenous group of disease at the molecular level. More importantly, it is now a disease with a genetic basis. The specific genetic signature that predisposes to its malignant progression is just being unraveled. From the clinical standpoint, it will be crucial for clinicians to define this signature in order to cater our treatment strategy to the risk of malignancy.
References
Flejou JF, Barge J, Menu Y, Degott C, Bismuth H, Potet F, Benhamou JP. Liver adenomatosis. An entity distinct from liver adenoma? Gastroenterology 1985;89(5): 1132–1138.
Bioulac-Sage P, Balabaud C, Bedossa P, Scoazec JY, Chiche L, Dhillon AP, Ferrell L, Paradis V, Roskams T, Vilgrain V, Wanless IR, Zucman-Rossi J. Pathological diagnosis of liver cell adenoma and focal nodular hyperplasia: Bordeaux update. Journal of hepatology 2007;46(3): 521–527.
van der Windt DJ, Kok NF, Hussain SM, Zondervan PE, Alwayn IP, de Man RA, JN IJ. Case-orientated approach to the management of hepatocellular adenoma. Br J Surg 2006; 93(12): 1495–1502.
Ball C, Sackett D, Phillips B, Haynes B, Straus S. Levels of evidence and grades of recommendations. In: EBM (Web site of the Oxford Centre for Evidence-Based Medicine); 2009, http://www.cebm.net/
Jeannot E, Wendum D, Paye F, Mourra N, de Toma C, Flejou JF, Zucman-Rossi J. Hepatocellular adenoma displaying a HNF1alpha inactivation in a patient with familial adenomatous polyposis coli. Journal of Hepatology 2006;45(6): 883–886.
Rooks JB, Ory HW, Ishak KG, Strauss LT, Greenspan JR, Hill AP, Tyler CW, Jr. Epidemiology of hepatocellular adenoma. The role of oral contraceptive use. Jama 1979;242(7): 644–648.
Lee PJ. Glycogen storage disease type I: pathophysiology of liver adenomas. Eur J Pediatr 2002;161 Suppl 1: S46-49.
Steinbrecher UP, Lisbona R, Huang SN, Mishkin S. Complete regression of hepatocellular adenoma after withdrawal of oral contraceptives. Dig Dis Sci 1981;26(11): 1045–1050.
Aseni P, Sansalone CV, Sammartino C, Benedetto FD, Carrafiello G, Giacomoni A, Osio C, Vertemati M, Forti D. Rapid disappearance of hepatic adenoma after contraceptive withdrawal. J Clin Gastroenterol 2001;33(3): 234–236.
Ramseur WL, Cooper MR. Asymptomatic liver cell adenomas. Another case of resolution after discontinuation of oral contraceptive use. Jama 1978;239(16): 1647–1648.
Edmondson HA, Reynolds TB, Henderson B, Benton B. Regression of liver cell adenomas associated with oral contraceptives. Ann Intern Med 1977;86(2): 180–182.
Torbenson M, Lee JH, Choti M, Gage W, Abraham SC, Montgomery E, Boitnott J, Wu TT. Hepatic adenomas: analysis of sex steroid receptor status and the Wnt signaling pathway. Mod Pathol 2002;15(3): 189–196.
Demo E, Frush D, Gottfried M, Koepke J, Boney A, Bali D, Chen YT, Kishnani PS. Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? Journal of hepatology 2007;46(3): 492–498.
Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr 1997;24(3): 276–279.
Bannasch P. Sequential cellular changes during chemical carcinogenesis. J Cancer Res Clin Oncol 1984;108(1): 11–22.
Bannasch P, Mayer D, Hacker HJ. Hepatocellular glycogenosis and hepatocarcinogenesis. Biochim Biophys Acta 1980;605(2): 217–245.
Paradis V, Laurent A, Flejou JF, Vidaud M, Bedossa P. Evidence for the polyclonal nature of focal nodular hyperplasia of the liver by the study of X-chromosome inactivation. Hepatology 1997;26(4): 891–895.
Franco LM, Krishnamurthy V, Bali D, Weinstein DA, Arn P, Clary B, Boney A, Sullivan J, Frush DP, Chen YT, Kishnani PS. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis 2005;28(2): 153–162.
Tao LC. Oral contraceptive-associated liver cell adenoma and hepatocellular carcinoma. Cytomorphology and mechanism of malignant transformation. Cancer 1991;68(2): 341–347.
Zucman-Rossi J, Jeannot E, Nhieu JT, Scoazec JY, Guettier C, Rebouissou S, Bacq Y, Leteurtre E, Paradis V, Michalak S, Wendum D, Chiche L, Fabre M, Mellottee L, Laurent C, Partensky C, Castaing D, Zafrani ES, Laurent-Puig P, Balabaud C, Bioulac-Sage P. Genotype-phenotype correlation in hepatocellular adenoma: new classification and relationship with HCC. Hepatology 2006;43(3): 515–524.
Micchelli ST, Vivekanandan P, Boitnott JK, Pawlik TM, Choti MA, Torbenson M. Malignant transformation of hepatic adenomas. Mod Pathol 2008;21(4): 491–497.
Tao LC. Are oral contraceptive-associated liver cell adenomas premalignant? Acta Cytol 1992;36(3): 338–344.
Anthony PP, Vogel CL, Barker LF. Liver cell dysplasia: a premalignant condition. J Clin Pathol 1973;26(3): 217–223.
Farges O, Ferreira N, Dokmak S, Belghiti J, Bedossa P, Paradis V. Changing trends in malignant transformation of hepatocellular adenoma. Gut 2011;60(1): 85–89.
Chiche L, Dao T, Salame E, Galais MP, Bouvard N, Schmutz G, Rousselot P, Bioulac-Sage P, Segol P, Gignoux M. Liver adenomatosis: reappraisal, diagnosis, and surgical management: eight new cases and review of the literature. Ann Surg 2000;231(1): 74–81.
Limmer J, Fleig WE, Leupold D, Bittner R, Ditschuneit H, Beger HG. Hepatocellular carcinoma in type I glycogen storage disease. Hepatology 1988;8(3): 531–537.
Kudo M. Hepatocellular adenoma in type Ia glycogen storage disease. J Gastroenterol 2001;36(1): 65–66.
Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004;432(7015): 324–331.
Libbrecht L, Desmet V, Van Damme B, Roskams T. The immunohistochemical phenotype of dysplastic foci in human liver: correlation with putative progenitor cells. Journal of hepatology 2000;33(1): 76–84.
Libbrecht L, De Vos R, Cassiman D, Desmet V, Aerts R, Roskams T. Hepatic progenitor cells in hepatocellular adenomas. The American journal of surgical pathology 2001;25(11): 1388–1396.
Deneve JL, Pawlik TM, Cunningham S, Clary B, Reddy S, Scoggins CR, Martin RC, D'Angelica M, Staley CA, Choti MA, Jarnagin WR, Schulick RD, Kooby DA. Liver cell adenoma: a multicenter analysis of risk factors for rupture and malignancy. Ann Surg Oncol 2009;16(3): 640–648.
Stoot JH, Coelen RJ, De Jong MC, Dejong CH. Malignant transformation of hepatocellular adenomas into hepatocellular carcinomas: a systematic review including more than 1600 adenoma cases. HPB : the official journal of the International Hepato Pancreato Biliary Association 2010;12(8): 509–522.
Bioulac-Sage P, Laumonier H, Sa Cunha A, Balabaud C. Hepatocellular adenomas. Liver Int 2009;29(1): 142.
Bioulac-Sage P, Rebouissou S, Thomas C, Blanc JF, Saric J, Sa Cunha A, Rullier A, Cubel G, Couchy G, Imbeaud S, Balabaud C, Zucman-Rossi J. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology 2007;46(3): 740–748.
Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular Benign Tumors-From Molecular Classification to Personalized Clinical Care. Gastroenterology 2013.
Rebouissou S, Imbeaud S, Balabaud C, Boulanger V, Bertrand-Michel J, Terce F, Auffray C, Bioulac-Sage P, Zucman-Rossi J. HNF1alpha inactivation promotes lipogenesis in human hepatocellular adenoma independently of SREBP-1 and carbohydrate-response element-binding protein (ChREBP) activation. J Biol Chem 2007;282(19): 14437–14446.
Van der Borght S, Libbrecht L, Katoonizadeh A, Aerts R, Nevens F, Verslype C, Roskams TA. Nuclear beta-catenin staining and absence of steatosis are indicators of hepatocellular adenomas with an increased risk of malignancy. Histopathology 2007;51(6): 855–856.
Wilkens L, Bredt M, Flemming P, Becker T, Klempnauer J, Kreipe HH. Differentiation of liver cell adenomas from well-differentiated hepatocellular carcinomas by comparative genomic hybridization. J Pathol 2001;193(4): 476–482.
Nasarek A, Werner M, Nolte M, Klempnauer J, Georgii A. Trisomy 1 and 8 occur frequently in hepatocellular carcinoma but not in liver cell adenoma and focal nodular hyperplasia. A fluorescence in situ hybridization study. Virchows Arch 1995;427(4): 373–378.
Wilkens L, Bredt M, Flemming P, Schwarze Y, Becker T, Mengel M, von Wasielewski R, Klempnauer J, Kreipe H. Diagnostic impact of fluorescence in situ hybridization in the differentiation of hepatocellular adenoma and well-differentiated hepatocellular carcinoma. J Mol Diagn 2001;3(2): 68–73.
De Souza AT, Hankins GR, Washington MK, Fine RL, Orton TC, Jirtle RL. Frequent loss of heterozygosity on 6q at the mannose 6-phosphate/insulin-like growth factor II receptor locus in human hepatocellular tumors. Oncogene 1995;10(9): 1725–1729.
Macdonald GA, Greenson JK, Saito K, Cherian SP, Appelman HD, Boland CR. Microsatellite instability and loss of heterozygosity at DNA mismatch repair gene loci occurs during hepatic carcinogenesis. Hepatology 1998;28(1): 90–97.
Bluteau O, Jeannot E, Bioulac-Sage P, Marques JM, Blanc JF, Bui H, Beaudoin JC, Franco D, Balabaud C, Laurent-Puig P, Zucman-Rossi J. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet 2002;32(2): 312–315.
Jeannot E, Poussin K, Chiche L, Bacq Y, Sturm N, Scoazec JY, Buffet C, Van Nhieu JT, Bellanne-Chantelot C, de Toma C, Laurent-Puig P, Bioulac-Sage P, Zucman-Rossi J. Association of CYP1B1 germ line mutations with hepatocyte nuclear factor 1alpha-mutated hepatocellular adenoma. Cancer Res 2007;67(6): 2611–2616.
Reznik Y, Dao T, Coutant R, Chiche L, Jeannot E, Clauin S, Rousselot P, Fabre M, Oberti F, Fatome A, Zucman-Rossi J, Bellanne-Chantelot C. Hepatocyte nuclear factor-1 alpha gene inactivation: cosegregation between liver adenomatosis and diabetes phenotypes in two maturity-onset diabetes of the young (MODY)3 families. J Clin Endocrinol Metab 2004;89(3): 1476–1480.
Bacq Y, Jacquemin E, Balabaud C, Jeannot E, Scotto B, Branchereau S, Laurent C, Bourlier P, Pariente D, de Muret A, Fabre M, Bioulac-Sage P, Zucman-Rossi J. Familial liver adenomatosis associated with hepatocyte nuclear factor 1alpha inactivation. Gastroenterology 2003;125(5): 1470–1475.
Foster JH, Donohue TA, Berman MM. Familial liver-cell adenomas and diabetes mellitus. N Engl J Med 1978;299(5): 239–241.
Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, Chen X, Cox NJ, Oda Y, Yano H, Le Beau MM, Yamada S, Nishigori H, Takeda J, Fajans SS, Hattersley AT, Iwasaki N, Hansen T, Pedersen O, Polonsky KS, Bell GI, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature 1996;384(6608): 455–458.
Micsenyi A, Tan X, Sneddon T, Luo JH, Michalopoulos GK, Monga SP. Beta-catenin is temporally regulated during normal liver development. Gastroenterology 2004;126(4): 1134–1146.
Monga SP, Monga HK, Tan X, Mule K, Pediaditakis P, Michalopoulos GK. Beta-catenin antisense studies in embryonic liver cultures: role in proliferation, apoptosis, and lineage specification. Gastroenterology 2003;124(1): 202–216.
Chen YW, Jeng YM, Yeh SH, Chen PJ. P53 gene and Wnt signaling in benign neoplasms: beta-catenin mutations in hepatic adenoma but not in focal nodular hyperplasia. Hepatology 2002;36(4 Pt 1): 927–935.
de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O, Fabre M, Chelly J, Beldjord C, Kahn A, Perret C. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A 1998;95(15): 8847–8851.
Miyoshi Y, Iwao K, Nagasawa Y, Aihara T, Sasaki Y, Imaoka S, Murata M, Shimano T, Nakamura Y. Activation of the beta-catenin gene in primary hepatocellular carcinomas by somatic alterations involving exon 3. Cancer Res 1998;58(12): 2524–2527.
Nhieu JT, Renard CA, Wei Y, Cherqui D, Zafrani ES, Buendia MA. Nuclear accumulation of mutated beta-catenin in hepatocellular carcinoma is associated with increased cell proliferation. Am J Pathol 1999;155(3): 703–710.
Laurent-Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, Monges G, Thomas G, Bioulac-Sage P, Zucman-Rossi J. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology 2001;120(7): 1763–1773.
Rebouissou S, Amessou M, Couchy G, Poussin K, Imbeaud S, Pilati C, Izard T, Balabaud C, Bioulac-Sage P, Zucman-Rossi J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009;457(7226): 200–204.
Bala S, Wunsch PH, Ballhausen WG. Childhood hepatocellular adenoma in familial adenomatous polyposis: mutations in adenomatous polyposis coli gene and p53. Gastroenterology 1997;112(3): 919–922.
Blaker H, Sutter C, Kadmon M, Otto HF, Von Knebel-Doeberitz M, Gebert J, Helmke BM. Analysis of somatic APC mutations in rare extracolonic tumors of patients with familial adenomatous polyposis coli. Genes Chromosomes Cancer 2004;41(2): 93–98.
Tannapfel A, Busse C, Geissler F, Witzigmann H, Hauss J, Wittekind C. INK4a-ARF alterations in liver cell adenoma. Gut 2002;51(2): 253–258.
Ladeiro Y, Couchy G, Balabaud C, Bioulac-Sage P, Pelletier L, Rebouissou S, Zucman-Rossi J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 2008;47(6): 1955–1963.
Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, Kay MA, Wang R, Bishop JM. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007;104(37): 14771–14776.
Pontoglio M, Barra J, Hadchouel M, Doyen A, Kress C, Bach JP, Babinet C, Yaniv M. Hepatocyte nuclear factor 1 inactivation results in hepatic dysfunction, phenylketonuria, and renal Fanconi syndrome. Cell 1996;84(4): 575–585.
Lee YH, Sauer B, Gonzalez FJ. Laron dwarfism and non-insulin-dependent diabetes mellitus in the Hnf-1alpha knockout mouse. Mol Cell Biol 1998;18(5): 3059–3068.
Flodby P, Liao DZ, Blanck A, Xanthopoulos KG, Hallstrom IP. Expression of the liver-enriched transcription factors C/EBP alpha, C/EBP beta, HNF-1, and HNF-4 in preneoplastic nodules and hepatocellular carcinoma in rat liver. Mol Carcinog 1995;12(2): 103–109.
Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Guidelines for management of glycogen storage disease type I-European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr 2002;161 Suppl 1: S112-119.
Lee SH, Hahn ST. Treatment of multiple hepatic adenomatosis using transarterial chemoembolization: a case report. Cardiovasc Intervent Radiol 2004;27(5): 563–565.
Akovbiantz A, Ackermann D, Buhler H, Schmid M. [Management of liver cell adenoma and focal nodular hyperplasia]. Helv Chir Acta 1980;47(5): 607–609.
Monga SP. Hepatic adenomas: presumed innocent until proven to be beta-catenin mutated. Hepatology 2006;43(3): 401–404.
Davis M, Portmann B, Searle M, Wright R, Williams R. Histological evidence of carcinoma in a hepatic tumor associated with oral contraceptives. Br Med J 1975;4(5995): 496–498.
Pryor AC, Cohen RJ, Goldman RL. Hepatocellular carcinoma in a woman on long-term oral contraceptives. Cancer 1977;40(2): 884–888.
Tesluk H, Lawrie J. Hepatocellular adenoma. Its transformation to carcinoma in a user of oral contraceptives. Arch Pathol Lab Med 1981;105(6): 296–299.
Gordon SC, Reddy KR, Livingstone AS, Jeffers LJ, Schiff ER. Resolution of a contraceptive-steroid-induced hepatic adenoma with subsequent evolution into hepatocellular carcinoma. Ann Intern Med 1986;105(4): 547–549.
Gyorffy EJ, Bredfeldt JE, Black WC. Transformation of hepatic cell adenoma to hepatocellular carcinoma due to oral contraceptive use. Ann Intern Med 1989;110(6): 489–490.
Korula J, Yellin A, Kanel G, Campofiori G, Nichols P. Hepatocellular carcinoma coexisting with hepatic adenoma. Incidental discovery after long-term oral contraceptive use. West J Med 1991;155(4): 416–418.
Ferrell LD. Hepatocellular carcinoma arising in a focus of multilobular adenoma. A case report. The American journal of surgical pathology 1993;17(5): 525–529.
Janes CH, McGill DB, Ludwig J, Krom RA. Liver cell adenoma at the age of 3 years and transplantation 19 years later after development of carcinoma: a case report. Hepatology 1993;17(4): 583–585.
Perret AG, Mosnier JF, Porcheron J, Cuilleron M, Berthoux P, Boucheron S, Audigier JC. Role of oral contraceptives in the growth of a multilobular adenoma associated with a hepatocellular carcinoma in a young woman. Journal of hepatology 1996;25(6): 976–979.
Ye MQ, Suriawinata A, Ben Haim M, Parsons R, Schwartz ME. A 42-year-old woman with liver masses and long-term use of oral contraceptives. Semin Liver Dis 1999;19(3): 339–344.
Burri E, Steuerwald M, Cathomas G, Mentha G, Majno P, Rubbia-Brandt L, Meier R. Hepatocellular carcinoma in a liver-cell adenoma within a non-cirrhotic liver. European journal of gastroenterology & hepatology 2006;18(4): 437–441.
Colovic R, Grubor N, Micev M, Radak V. Hepatocellular adenoma with malignant alteration. Hepatogastroenterology 2007;54(74): 386–388.
Kim DH, Kim SU, Nam DH, Choi YJ, Park SM, Lee CK, Kim do Y. A case of hepatocellular carcinoma within hepatocellular adenoma in a non-cirrhotic male. The Korean journal of internal medicine 2009;24(2): 147–152.
Toiyama Y, Inoue Y, Yasuda H, Yoshiyama S, Araki T, Miki C, Kusunoki M. Hepatocellular adenoma containing hepatocellular carcinoma in a male patient with familial adenomatous polyposis coli: Report of a case. Surgery today 2011;41(10): 1442–1446.
Hechtman JF, Raoufi M, Fiel MI, Taouli B, Facciuto M, Schiano TD, Blouin AG, Thung SN. Hepatocellular carcinoma arising in a pigmented telangiectatic adenoma with nuclear beta-catenin and glutamine synthetase positivity: case report and review of the literature. The American journal of surgical pathology 2011;35(6): 927–932.
Lack EE, Neave C, Vawter GF. Hepatocellular carcinoma. Review of 32 cases in childhood and adolescence. Cancer 1983;52(8): 1510–1515.
Kerlin P, Davis GL, McGill DB, Weiland LH, Adson MA, Sheedy PF, 2nd. Hepatic adenoma and focal nodular hyperplasia: clinical, pathologic, and radiologic features. Gastroenterology 1983;84(5 Pt 1): 994–1002.
Leese T, Farges O, Bismuth H. Liver cell adenomas. A 12-year surgical experience from a specialist hepato-biliary unit. Ann Surg 1988;208(5): 558–564.
Belghiti J, Pateron D, Panis Y, Vilgrain V, Flejou JF, Benhamou JP, Fekete F. Resection of presumed benign liver tumours. Br J Surg 1993;80(3): 380–383.
Herman P, Machado MA, Volpe P, Pugliese V, Vianna MR, Bacchella T, Machado MC, Pinotti HW. [Transformation of hepatic adenoma into hepatocellular carcinoma in patients with prolonged use of oral contraceptives]. Rev Hosp Clin Fac Med Sao Paulo 1994;49(1): 30–33.
Foster JH, Berman MM. The malignant transformation of liver cell adenomas. Arch Surg 1994;129(7): 712–717.
Nagorney DM. Benign hepatic tumors: focal nodular hyperplasia and hepatocellular adenoma. World journal of surgery 1995;19(1): 13–18.
Ault GT, Wren SM, Ralls PW, Reynolds TB, Stain SC. Selective management of hepatic adenomas. Am Surg 1996;62(10): 825–829.
De Carlis L, Pirotta V, Rondinara GF, Sansalone CV, Colella G, Maione G, Slim AO, Rampoldi A, Cazzulani A, Belli L, Forti D. Hepatic adenoma and focal nodular hyperplasia: diagnosis and criteria for treatment. Liver Transpl Surg 1997;3(2): 160–165.
Weimann A, Ringe B, Klempnauer J, Lamesch P, Gratz KF, Prokop M, Maschek H, Tusch G, Pichlmayr R. Benign liver tumors: differential diagnosis and indications for surgery. World journal of surgery 1997;21(9): 983–990; discussion 990–981.
Closset J, Veys I, Peny MO, Braude P, Van Gansbeke D, Lambilliotte JP, Gelin M. Retrospective analysis of 29 patients surgically treated for hepatocellular adenoma or focal nodular hyperplasia. Hepatogastroenterology 2000;47(35): 1382–1384.
Ichikawa T, Federle MP, Grazioli L, Nalesnik M. Hepatocellular adenoma: multiphasic CT and histopathologic findings in 25 patients. Radiology 2000;214(3): 861–868.
Reddy KR, Kligerman S, Levi J, Livingstone A, Molina E, Franceschi D, Badalamenti S, Jeffers L, Tzakis A, Schiff ER. Benign and solid tumors of the liver: relationship to sex, age, size of tumors, and outcome. Am Surg 2001;67(2): 173–178.
Charny CK, Jarnagin WR, Schwartz LH, Frommeyer HS, DeMatteo RP, Fong Y, Blumgart LH. Management of 155 patients with benign liver tumours. Br J Surg 2001;88(6): 808–813.
Marini P, Vilgrain V, Belghiti J. Management of spontaneous rupture of liver tumours. Digestive surgery 2002;19(2): 109–113.
Dokmak S, Paradis V, Vilgrain V, Sauvanet A, Farges O, Valla D, Bedossa P, Belghiti J. A single-center surgical experience of 122 patients with single and multiple hepatocellular adenomas. Gastroenterology 2009;137(5): 1698–1705.
Sasaki M, Yoneda N, Kitamura S, Sato Y, Nakanuma Y. Characterization of hepatocellular adenoma based on the phenotypic classification: The Kanazawa experience. Hepatology research : the official journal of the Japan Society of Hepatology 2011;41(10): 982–988.
Bellamy CO, Maxwell RS, Prost S, Azodo IA, Powell JJ, Manning JR. The value of immunophenotyping hepatocellular adenomas: consecutive resections at one UK centre. Histopathology 2013;62(3): 431–445.
Fonseca S, Hoton D, Dardenne S, Annet L, Hubert C, Godecharles S, Jouret-Mourin A, Reding R, Otte JB, Rahier J, Gigot JF, Sempoux C. Histological and immunohistochemical revision of hepatocellular adenomas: a learning experience. International journal of hepatology 2013;2013: 398308.
Acknowledgments
S-S Liau is in receipt of the MRC Clinician Scientist Fellowship. He is also funded by University of Cambridge Parke-Davis Fellowship, Royal Society of Medicine Ellison-Cliffe Fellowship, Dowager Countess Eleanor Peel Fellowship, HCA International Foundation Fellowship, European Society of Surgical Oncology Minor Fellowship, and MRC Centenary Early Career Award.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 161 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Liau, SS., Qureshi, M.S., Praseedom, R. et al. Molecular Pathogenesis of Hepatic Adenomas and Its Implications for Surgical Management. J Gastrointest Surg 17, 1869–1882 (2013). https://doi.org/10.1007/s11605-013-2274-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-013-2274-6