
Abstract
In recent years, a large number of clandestinely synthesized new psychoactive substances with high structural variety have been detected in forensic samples. Analytical differentiation of regioisomers is a significant issue in forensic drug analysis, because, in most cases, legal controls are placed on only one or two of the conceivable isomers. In this study, gas chromatography–tandem mass spectrometry (GC–MS–MS) was used to differentiate the regioisomers of chloroamphetamine analogs (chloroamphetamines and chloromethamphetamines) synthesized in the authors′ laboratories. Free bases, trifluoroacetyl derivatives, and trimethylsilyl derivatives were subjected to GC–MS–MS using DB-1ms, DB-5ms, and DB-17ms capillary columns, respectively. The regioisomers of chloroamphetamine analogs in all forms were well separated on the DB-5ms column. The electron ionization mass spectra of the chloroamphetamine analogs gave very little structural information for differentiation among these analogs, even after trifluoroacetyl and trimethylsilyl derivatization of the analytes. Characteristic product ions of the 2-positional isomers were observed by electron ionization-MS–MS. In contrast, chemical ionization-MS–MS of the free bases provided more structural information about chloride position on the aromatic ring when [M+H–HCl]+ was selected as a precursor ion. The results suggest that a combination of chromatographic analysis and MS–MS supports differentiation for regioisomers of chloroamphetamine analogs.
Similar content being viewed by others
Introduction
In recent years, a large number of controlled substance analogs with high structural variety have been widely distributed as noncontrolled alternatives and have been detected in forensic samples. Because of the frequent appearance of new analogs with displacement of a functional group or an alkyl chain [1–3], structural differentiation of these new drugs is necessary in forensic laboratories for legal control against drug abuse [4–14].
4-Chloromethamphetamine (4-CMA) was first reported as a selective serotoninergic depleting agent in rat brains in 1963 by Pletsher et al. [15]. Chlorinated analogs of amphetamine were subsequently synthesized and examined in the Lilly research laboratory as appetite depressants [16, 17]. It was eventually concluded that these analogs were not clinically useful and produced less central-nervous system stimulation than amphetamine or methamphetamine. After these reports were published, many researchers studied chloroamphetamines (CAPs) as factors of selective serotonin (5-hydroxytriptamine, 5-HT) diminution and as monoamine oxidase (MAO) inhibitors in brain nerve terminals [18–20]. It was also reported that 4-CAP and 4-CMA cause long-lasting depletion in the level of brain 5-HT and 5-hydroxyindole-3-acetic acid without depleting brain noradrenaline [21, 22]. Administration of 4-CAP to rats induced tremors, rigidity, Straub tail, hind limb abduction, lateral head weaving, and reciprocal forepaw treading [23]. It was previously reported that the half-life of 4-CAP was longer than that of 4-fluoroamphetamine (4-FAP) in rat brains, and that the liposolubility of 4-CAP was higher than that of 4-FAP in vitro [24]. Because of a tendency to dissolve in the organic phase, the depletion of brain 5-HT with 4-CAP continued for at least a week [24].
In Taiwan, CAP first appeared in April 2009, and a total of 775 cases were identified up to 2011 [25]. 4-CAP was also detected in urine samples in March 2010 [26]. In Hungary, it was detected in December 2012 as a new psychoactive substance and reported to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) and Europol for the first time [1]. Although there have been no reports of chloroamphetamine analogs (CAPs and CMAs) being abused and detected in forensic samples in Japan, 4-CAP has been controlled as a designated substance since August 2014.
For forensic purposes, differentiation of the chloro group ring position in amphetamine and methamphetamine is required because 4-CAP is controlled but 2-CAP and 3-CAP are not (Fig. 1). Although the results of infrared spectroscopy and nuclear magnetic resonance (NMR) spectrometry can provide structural information for identification of a specific isomer, these methods are not suitable for biological samples such as urine and blood because of their low sensitivity.

Chemical structures of chloroamphetamine analog regioisomers
Gas chromatography–mass spectrometry (GC–MS) is generally used for the identification of drugs because it is usually supported by an extensive electron ionization-mass spectrometry (EI-MS) database.
In this study, differentiation of chloroamphetamine analog regioisomers was studied using GC–MS–MS with EI and chemical ionization (CI). The effectiveness of analyte derivatization was also evaluated.
Materials and methods
Chemicals
4-Chlorophenylacetone, methylamine hydrochloride, and sodium cyanoborohydride were purchased from Tokyo Chemical Industry (Tokyo, Japan). Ortho- and m-chlorophenylacetone and trifluoroacetic anhydride (TFAA) were purchased from Wako Pure Chemical Industries (Osaka, Japan). N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) with 1 % trimethylchlorosilane (TMCS) was obtained from Supelco (St Louis, MO, USA). All other chemicals were of analytical grade.
Chloroamphetamine synthesis
Chlorophenylacetone and ammonium acetate or methylamine hydrochloride (5 equiv.) were dissolved in methanol at room temperature and the solution was stirred for 1 h. Sodium cyanoborohydride (0.4 equiv.) at 0 °C was added to the mixture. After the reaction was complete (Fig. 2), the pH of the mixture was adjusted to 12 with a 10 % potassium hydroxide aqueous solution. The mixture was extracted with diethyl ether, washed with water, and dried over sodium sulfate. The extract was then evaporated under reduced pressure. The crude residue was purified by column chromatography (silica gel, chloroform:methanol = 10:1).

Synthesis of chloroamphetamine analog regioisomers
Chemical structures were elucidated using 1H NMR spectroscopy. 1H NMR spectra were measured in deuterochloroform (CDCl3) with a JNM-ECP400 spectrometer (JEOL, Akishima, Japan) at room temperature. Tetramethylsilane was used as an internal standard. The 1H NMR data for each synthesized compound are summarized below.
-
2-CAP; 1H NMR (CDCl3) δ: 7.37–7.35 (1H, m), 7.22–7.14 (3H, m), 3.32–3.24 (1H, m), 2.86 (1H, dd, J = 13.2, 6.0 Hz), 2.68 (1H, dd, J = 13.2, 8.0 Hz), 1.14 (3H, d, J = 6.4 Hz).
-
3-CAP; 1H NMR (CDCl3) δ: 7.21–7.18 (3H, m), 7.08–7.06 (1H, m), 3.21–3.13 (1H, m), 2.68 (1H, dd, J = 13.2, 5.6 Hz), 2.51 (1H, dd, J = 13.2, 8.0 Hz), 1.12 (3H, d, J = 6.4 Hz).
-
4-CAP; 1H NMR (CDCl3) δ: 7.28–7.26 (2H, m), 7.13–7.10 (2H, m), 3.19–3.11 (1H, m), 2.67 (1H, dd, J = 13.6, 5.6 Hz), 2.52 (1H, dd, J = 13.2, 7.6 Hz), 1.05 (3H, d, J = 6.4 Hz).
-
2CMA; 1H NMR (CDCl3) δ: 7.36–7.34 (1H, m), 7.23–7.13 (3H, m), 2.97–2.87 (2H, m), 2.71–2.64 (1H, m), 2.44 (3H, s), 1.05 (3H, d, J = 6.4 Hz).
-
3-CMA; 1H NMR (CDCl3) δ: 7.24–7.18 (3H, m), 7.10–7.06 (1H, m), 2.83–2.69 (2H, m), 2.60–2.55 (1H, m), 2.40 (3H, s), 1.05 (3H, d, J = 5.6 Hz).
-
4-CMA; 1H NMR (CDCl3) δ: 7.27–7.25 (2H, m), 7.13–7.11 (2H, m), 2.83–2.70 (2H, m), 2.59 (1H, dd, J = 13.2, 6.4 Hz), 2.39 (3H, s), 1.05 (3H, d, J = 5.6 Hz).
Preparation of sample solution
Stock standard solutions of all six compounds as corresponding hydrochloride salts were prepared in distilled water to give a concentration of 1.0 mg/ml. The solutions were further diluted with distilled water to give appropriate concentrations.
Extraction procedure
One milliliter of 0.5 mg/ml aqueous solution containing hydrochloride salts of chloroamphetamine analogs was alkalinized with 20 μl of 2 M sodium hydroxide aqueous solution before being shaken with 1 ml of ethyl acetate for 3 min. After centrifugation, a portion of the extract was diluted by a factor of ten. The resulting solution was subjected to GC–MS.
Trifluoroacetylation
Up to 40 μl of ethyl acetate extract was evaporated to dryness under a gentle stream of nitrogen. The residue was dissolved in 200 μl of ethyl acetate and 200 μl of TFAA, and the mixture was heated for 15 min at 50 °C. After being cooled to room temperature, the mixture was evaporated to dryness under a gentle stream of nitrogen. The residue was redissolved in 1000 μl of ethyl acetate and subjected to GC–MS.
Trimethylsilylation
Ten microliters of extracted ethyl acetate solution was evaporated to dryness under a gentle stream of nitrogen. The dry residue was dissolved in 100 μl of acetonitrile, and 100 μl of BSTFA with 1 % TMCS content was added to the solution. The mixture was heated for 30 min at 80 °C and subjected to GC–MS analysis after being cooled to room temperature.
GC–MS conditions
GC–MS was performed on a 7000 Triple quadrupole GC–MS system (Agilent, Santa Clara, CA, USA) equipped with a DB-1ms, a DB-5ms, or a DB-17ms capillary column (30 m × 0.25 mm I.D., film thickness 0.25 μm, Agilent J&W, Folsom, CA, USA). The oven temperature was maintained at 80 °C for 1 min following injection and then raised to 300 °C at a rate of 15 °C/min. The injection port and interface temperature were set at 250 °C. Helium was used as the carrier gas at a flow rate of 1.0 ml/min. One microliter of sample solution was injected in splitless mode.
For EI mode, the ionization energy was 70 eV, the ion source temperature was 230 °C, and the scan mass range was m/z 40–400. For CI mode, the ionization energy was 70 eV, the ion source temperature was 250 °C, and the scan mass range was m/z 43–400.
Results and discussion
Gas chromatography
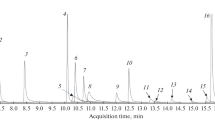
Figure 3 shows total ion current chromatograms of free bases, trifluoroacetyl (TFA) derivatives, and trimethylsilyl (TMS) derivatives of chloroamphetamine analogs. Retention indices of the analogs are shown in Table 1. The regioisomers of chloroamphetamine analogs in all the forms of free bases, TFA derivatives, and TMS derivatives were eluted in the order of 2-positional isomers > 3-positional isomers > 4-positional isomers. Free bases of six analytes were well separated from each other on the DB-5ms column, although the peak shapes showed tailing (Fig. 3b). Their TFA and TMS derivatives were separated completely with symmetric peaks on all three separation columns.

Total ion current chromatograms for free bases, trifluoroacetyl (TFA) derivatives, and trimethylsilyl (TMS) derivatives of chloroamphetamine analogs separated on DB-1ms (a), DB-5ms (b), and DB-17ms (c) columns. The compound names are the same as those in Fig. 1
Electron ionization-mass spectrometry
The EI mass spectra of free bases of chloroamphetamine analogs are shown in Fig. 4. The mass spectra of the isomers were very similar. Chloroamphetamine analogs showed the same major fragment ions based on benzyl bond cleavage (m/z 125) and α-cleavage (m/z 44 or 58). Their TFA and TMS derivatives also gave very little structural information for differentiation among these analogs (data not shown).

Electron ionization (EI) mass spectra for free bases of chloroamphetamine analogs
Tandem MS was applied under various conditions of collision energy. The characteristic fragmentation of 2-positional isomers was observed clearly from the product ion spectra of chloroamphetamine analogs with moderate collision energy (CE = 30, Fig. 5). The fragment ions at m/z 118 and 132 based on desorption of hydrochloride came from precursor ions at m/z 154 and 168. Meanwhile, 3- and 4-positional isomers produced fragment ions at m/z 119 and 133 based on chloride radical desorption from precursor ions rather than fragment ions of hydrochloride loss under the same collision energy. The loss of hydrochloride by the 2-positional Cl group on an aromatic ring in tandem MS was in accordance with the content of a previous report by Katiaho et al. [27]. The ratio of the fragment ion at m/z 42 for 3-CMA was higher than that for 4-CMA. The difference between 3-CMA and 4-CMA was more obvious because of strong collision energy (data not shown). The results obtained here suggest that careful analysis of tandem EI-MS with changes in collision energy supports the differentiation of CMA regioisomers.

EI product ion spectra for free bases of chloroamphetamines (CAPs) (precursor ions at m/z 154; a) and chloromethamphetamines (CMAs) (precursor ions at m/z 168; b)
TMS derivatives of 2-positional isomers showed a significant number of fragment ions at m/z 132 and 190 for 2-CAP and m/z 147 and 204 for 2-CMA in product ion spectra (Fig. 6). However, the spectra for TMS derivatives of 3- and 4-positional isomers were almost equivalent. It was considered that the product ions at m/z 132 and 147 were produced by desorption of [Si(CH3)2–Cl] from the precursor ion, [M–CH3]+. The ions at m/z 190 and 204 came from hydrochloride desorption of the precursor ions.

EI product ion spectra for TMS derivatives of CAPs (precursor ions at m/z 226, [M–CH3]+; a) and CMAs (precursor ions at m/z 240, [M–CH3]+; b)
Chemical ionization-mass spectrometry
The CI mass spectra for free bases of chloroamphetamine analogs are shown in Fig. 7. CI-MS of CAPs provided the same major fragment ions at m/z 153 and 170. In the spectra of CMAs, [M+H]+ at m/z 184 was also produced as the base peak. However, the mass spectra of 2-positional isomers were different from those of 3- and 4-positional isomers, the spectra of which were almost the same. Their fragment ion desorption of chloride ions (m/z 134 or 148) was also higher than that of benzyl cations (m/z 125). The 2-positional isomers produced an equivalent or higher ratio of benzyl cations than fragment ion produced by desorption of chloride ions. TFA and TMS derivatives were examined, but only TMS derivatives of 2-CAP were differentiated by a characteristic ion at m/z 206, [M+H–HCl]+ (data not shown).

Chemical ionization (CI) mass spectra for free bases of chloroamphetamine analogs
In contrast, product ion analysis of the free bases provided more structural information about chloride position on the aromatic ring when an optimal precursor ion was selected. Figure 8a shows the product ion spectra of CAP free bases obtained from the precursor ions at m/z 134, corresponding to [M+H–HCl]+. Three CAP isomers showed different spectrum patterns. In particular, 2-CAP was markedly different from the other isomers. Only 2-CAP gave the ions at m/z 132 (neutral loss of hydrogen molecule), 106, and 93. In addition, 2-CAP did not give the ions at m/z 44 (α-cleavaged iminium cation) and 91 (tropylium cation), which were observed for both 3-CAP and 4-CAP. These differences suggested that the precursor ions at m/z 134 in the CI mass spectra were composed of different chemical structures between 2-CAP and the others. The precursor ion of 2-CAP was presumed to be 2-methylindolinium ion (Fig. 9a). This structure was supported by the ion at m/z 106 in the product ion spectrum because this ion corresponded to anchimeric-stabilized amine tropylium cation generated by rearrangement with loss of ethylene from 2-methylindolium ion. This pathway was as reported in a previous study by Westphal et al. [28] on the differentiation of fluorinated amphetamines using GC–CI-MS–MS.

CI product ion spectra for free bases of CAPs (precursor ions at m/z 134, [M+H–HCl]+; a) and CMAs (precursor ions at m/z 148, [M+H–HCl]+; b)

Plausible structures of the precursor ions for CI–tandem mass spectrometry of CAPs and CMAs and their cleavage pathways. The precursor ions were m/z 134 for CAPs and m/z 148 for CMAs, which corresponded to [M+H–HCl]+
The ions at m/z 91 and 44 in the product ion spectra of 3-CAP and 4-CAP suggested that their precursor ions included the cations that were produced by simple elimination of HCl (Fig. 9b). These cations were cleaved to tropylium cation and iminium cation in product ion analysis (Fig. 9b). However, the ratio of m/z 91 against m/z 44 in 4-CAP was higher than that in 3-CAP, and their ratios were highly reproducible with a relative standard deviation (RSD) of less than 2 %. Moreover, the ion at m/z 119 was observed only in the product ion spectrum of 4-CAP. These findings suggested that the ions at m/z 134 in the CI spectra of 4-CAP was partially derived from a para-cyclophane-like cation (Fig. 9c). The ion at m/z 119 in the product ion spectrum of 4-CAP was produced by loss of methyl cation from the para-cyclophane-like cation. Moreover, considering the difference in the ratio of m/z 91 and 44 between 4-CAP and 3-CAP, the tropylium cation of 4-CAP may be partially derived from the para-cyclophane-like cation.
Product ion spectra of CMAs differed between isomers with the same tendency as CAPs. Figure 8b shows the product ion spectra of CMA free bases obtained from the precursor ions at m/z 148, corresponding to [M+H–HCl]+. 2-CMA showed the ions at m/z 146, 132, and 107, which were not observed in the spectra of 3-CMA and 4-CMA. 2-CMA did not give the ions at m/z 58 (α-cleavaged iminium cation) and 91 (tropylium cation), which were observed for both 3-CMA and 4-CMA. The ratios of m/z 91 and 58 were different between 3-CMA and 4-CMA with high reproducibility (RSD less than 2 %). 4-CMA gave a characteristic ion of m/z 133, corresponding to the loss of methyl cation from the precursor ion. These findings suggested the ions at m/z 148 in the CI spectra originated from 2-methylindolinium for 2-CMA (Fig. 9d), a cation which was produced by simple elimination of HCl for 3-CMA (Fig. 9e), and the simply eliminated cation and para-cyclophane-like cation for 4-CMA (Fig. 9e, f).
The CI product ion spectra of TMS derivatives of 2-positional isomers were clearly different from those of 3- and 4-positional isomers (Fig. 10). In contrast, the CI-MS–MS of TFA derivatives gave almost the same spectra (data not shown). These results indicate that product ion spectrometry with optimal precursor ions based on CI-MS–MS can be used to differentiate between chloroamphetamine analogs.

CI product ion spectra for TMS derivatives of CAPs (precursor ions at m/z 226, [M–CH3]+; a) and CMAs (precursor ions at m/z 240, [M–CH3]+; b)
Conclusions
Six ring-substituted chloroamphetamine analogs were successfully separated using GC–MS with DB-1ms, DB-5ms, and DB-17ms columns. EI-MS provided structural information corresponding to the position of a chloride atom on the aromatic ring. All regioisomers of chloroamphetamine analogs as free bases could be differentiated using CI-MS–MS on [M+H–HCl]+. The results suggest that a combination of chromatographic analysis and tandem MS allow the differentiation of regioisomers of chloroamphetamine analogs.
References
EMCDDA-Europol (2012) Annual report on the implementation of council decision. 2005/387/JHA. doi: 10.2810/99367 (http://www.emcdda.europa.eu/attachements.cfm/att_212366_EN_EMCDDA-Europol%202012%20Annual%20Report_final.pdf)
INCB. annual report (2013) New psychoactive substances and other non-scheduled chemicals represent a clear and present danger. https://www.incb.org/documents/Publications/AnnualReports/AR2013/English/AR_2013_E.pdf
EU drug market report. strategic analysis. doi:102.2810/85143 (http://www.emcdda.europa.eu/attachements.cfm/att_194336_EN_TD3112366ENC.pdf)
Pirisi MA, Nieddu M, Burrai L, Carta A, Briguglio I, Baralla E, Demontis MP, Varoni MV, Boatto G (2013) An LC–MS–MS method for quantitative analysis of six trimethoxyamphetamine designer drugs in rat plasma, and its application to a pharmacokinetic study. Forensic Toxicol 31(2):197–203
Nakazono Y, Tsujikawa K, Kuwayama K, Kanamori T, Iwata YT, Miyamoto K, Kasuya F, Inoue H (2013) Differentiation of regioisomeric fluoroamphetamine analogs by gas chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry. Forensic Toxicol 31:241–250
Zaitsu K, Miyagawa H, Sakamoto Y, Matsuta S, Tsuboi K, Nishioka H, Katagi M, Sato T, Tatsuno M, Tsuchihashi H, Suzuki K, Ishii A (2013) Mass spectrometric differentiation of the isomers of mono-methoxyethylamphetamines and mono-methoxydimethylamphetamines by GC–EI-MS–MS. Forensic Toxicol 31:292–300
Nakazono Y, Tsujikawa K, Kuwayama K, Kanamori T, Iwata YT, Miyamoto K, Kasuya F, Inoue H (2014) Simultaneous determination of tryptamine analogs in designer drugs using gas chromatography–mass spectrometry and liquid chromatography–tandem mass spectrometry. Forensic Toxicol 32:154–161
Negishi S, Nakazono Y, Tsujikawa K, Kuwayama K, Kanamori T, Iwata YT, Miyamoto K, Kasuya F, Inoue H (2014) Differentiation of regioisomeric methylamphetamines using GC/MS (in Japanese). Jpn J Forensic Sci Tech 19:111–119
Belal T, Awad T, DeRuiter Clark CR (2008) GC-MS studies on acylated derivatives of 3-methoxy-4-methyl- and 4-methoxy-3-methyl-phenethylamines: regioisomers related to 3,4-MDMA. Forensic Sci Int 178:61–82
Clark CR, DeRuiter J (1996) Chromatographic and mass spectrometric methods for the differentiation of N-methyl-1-(3,4-methylendioxyphenyl)-2-butanomine from regioisomeric derivatives. J Chromatogr Sci 34:230–237
Borth S, Hansel W, Rosner P, Junge T (2000) Synthesis of 2,3- and 3,4-methylendioxyphenylalkylamines and their regioisomeric differentiation by mass spectral analysis using GC-MS-MS. Forensic Sci Int 114:139–153
Zaitsu K, Katagi M, Kamata HT, Miki A, Tsuchihashi H (2008) Discrimination and identification of regioisomeric β-keto analogues of 3,4-methylendioxyamphetamines by gas chromatography–mass spectrometry. Forensic Toxicol 26:45–51
Zaitsu K, Katagi M, Kamata H, Kamata T, Shima N, Miki A, Iwamura T, Tsuchihashi H (2008) Discrimination and identification of the six aromatic positional isomers of trimethoxyamphetamine (TMA) by gas chromatography-mass spectrometry (GC-MS). J Mass Spectrom 43:528–534
Kusano M, Zaitsu K, Nakayama H, Nakajima J, Hisatsune K, Moriyasu T, Matsuta S, Katagi M, Tsuchihashi H, Ishii A (2015) Positional isomer differentiation of synthetic cannabinoid JWH-081 by GC-MS/MS. J Mass Spectrom 50:586–591
Pletsher A, Burkhard WP, Bruderer H, Gey KF (1963) Decrease of cerebral 5-hydroxytryptamine and 5-hydroxyindoleacetic acid by an arylalkylamine. Life Sci 2:828–833
Owen JE Jr (1963) Psychopharmacological studies of some 1-(chlorophenyl)-2-aminopropanes. I. Effects on appetite-controlled behavior. J Pharm Sci 52:679–683
Owen JE Jr (1963) Psychopharmacological studies of some 1-(chlorophenyl)-2-aminopropanes. II. Effects on avoidance and discrimination behavior. J Pharm Sci 52:684–688
Harvey JA, McMaster SE, Yunger LM (1975) p-Chloroamphetamine: selective neurotoxic action in brain. Science 187:841–843
Ögren SO, Ross SB (1977) Substituted amphetamine derivatives. II. Behavioral effects in mice related to monoaminergic neurones. Acta Pharmacol Toxicol 41:353–368
Fuller RW (1992) Effects of p-chloroamphetamine on brain serotonin neurons. Neurochem Res 17:449–456
Miller FP, Cox RH Jr, Snodgrass WR, Maickel RP (1970) Comparative effects of p-chlorophenylalanine, p-chloroamphetamine and p-chloro-N-methylamphetamine on rat brain norepinephrine, serotonin and 5-hydroxyindole-3-acetic acid. Biochem Pharmacol 19:435–436
Sanders-Bush E, Sulser F (1970) p-Chloroamphetamine: in vivo investigations on the mechanism of action of the selective depletion of cerebral serotonin. J Pharm Exp Ther 175:419–426
Trulson ME, Jacobs BL (1976) Behavioral evidence for the rapid release of CNS serotonin by PCA and fenfluramine. Eur J Pharmacol 36:149–154
Fuller RW, Baker KW, Perry KW, Molloy BB (1975) Comparison of 4-chloro-, 4-bromo and 4-fluoroamphetamine in rats: drug levels in brain and effects on brain serotonin metabolism. Neuropharmacology 14:739–746
Lee SF, Hsu J, Tsay WI (2013) The trend of drug abuse in Taiwan during the years 1999 to 2011. J Food Drug Anal 21:390–396
Lin TC, Lin DL, Lua AC (2011) Detection of p-chloroamphetamine in urine samples with mass spectrometry. J Anal Toxicol 35:205–210
Kotiaho T, Shay BJ, Cooks RG, Eberlin MN (1993) Electrophilic aromatic Cl+ addition and CO∙+ substitution in the gas phase. J Am Chem Soc 115:1004–1014
Westphal F, Rösner P, Junge Th (2010) Differentiation of regioisomeric ring-substituted fluorophenethylamines with product ion spectrometry. Forensic Sci Int 194:53–59
Acknowledgments
This work was supported in part by the R&D Program for Implementation of Anti-Crime and Anti-Terrorism Technologies for a Safe and Secure Society and by Funds for Integrated Promotion of Social System Reform and Research and Development (provided by Japan’s Ministry of Education, Culture, Sports, Science and Technology).
Conflict of interest
There are no financial or other considerations that could lead to a conflict of interest in relation to this study.
Ethical approval
This article does not contain any studies with human participants or animals.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Negishi, S., Nakazono, Y., Iwata, Y.T. et al. Differentiation of regioisomeric chloroamphetamine analogs using gas chromatography–chemical ionization-tandem mass spectrometry. Forensic Toxicol 33, 338–347 (2015). https://doi.org/10.1007/s11419-015-0280-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-015-0280-y