
Abstract
Sixty-two strains of avian infectious bronchitis virus (IBV) were isolated from diseased chickens at different farms in southern China during 2011–2012, and 66.1 % of the isolated strains were associated with typical nephritis. Analysis of the S1 gene sequences amplified from the 62 isolated strains together with 40 reference strains published in Genbank showed nucleotide homologies ranging from 63.5 to 99.9 % and amino acid homologies ranging from 57.9 to 100 %. Phylogenetic analysis revealed that all Chinese IBV strains were clustered into six distinct genetic groups (I–VI). Most of the isolated strains belonged to group I, and the isolation of group V strains was increased compared with an earlier period of surveillance. Current vaccine strains used in China (H120, H52, W93, and Ma5) formed the group Mass which is evolutionarily distant from Chinese isolates. Alignment of S1 amino acid sequences revealed polymorphic and diverse substitutions, insertions, and deletions, and the S1 protein of major pandemic strains contained 540 amino acids with a cleavage site sequence of HRRRR or RRF(L/S)RR. Further analysis showed that recombination events formed a new subgroup. Taken together, these findings suggest that various IBV variants were co-circulating and undergoing genetic evolution in southern China during the observation period. Therefore, long-term continuing surveillance is significantly important for prevention and control of IBV infection.
Similar content being viewed by others

Introduction
Avian infectious bronchitis (IB) is an acute and highly contagious disease caused by the avian infectious bronchitis virus (IBV). It has a major impact on the poultry industry worldwide and causes severe economic losses [1].
IBV belongs to the genus Coronaviridae of the family Coronaviridae in the order Nidovirales. The IBV genome consists of a linear, non-segmented, positive-sense, single-stranded RNA of approximately 27.6 kilobases (kb) in length, encoding four structural proteins: nucleocapsid (N) protein, envelope protein (E), membrane glycoprotein (M), and spike glycoprotein (S). The spike glycoprotein is translated as a precursor protein (S0) and later cleaved into the S1 and S2 subunits by cellular proteases [2]. The S1 subunit forms the tip of a spike, and the S2 subunit anchors the S1 to the viral membrane. The S1 subunit, containing virus neutralizing epitopes and serotype-specific sequences [3–5], plays an important role in attachment to the host cell, as well as induction of neutralizing antibodies [6, 7]. Therefore, the genetic analysis of IBV is mainly based on the S1 gene [5, 8].
The continuous emergence of variant strains of IBV has been reported, and at least 30 serotypes have been identified worldwide. Some studies have indicated that IBV immunity is serotype-specific, and small differences in S1 protein may contribute to significant divergence in serotype as well as poor cross-protection [9, 10]. Three major factors account for the genetic diversity of IBV. First, the inaccuracy of the coronavirus RNA-dependent RNA polymerase (RDRP) results in continuous evolution of the virus through point mutations, insertions, and deletions [11]. Second, IBV undergoes a high frequency of homologous RNA recombination due to its unique random template switching during RNA replication [12]. Third, immune pressure is exerted on circulating viruses by the constant presence of partially immune chicken populations. These three factors have not only led to a diversity of strains and genotypes, but also generated new species that can adapt to new hosts and ecological niches [13]. The design of appropriate control programs has been complicated by the large number of IBV serotype or genotypes [14], as well as the low degree of cross-protection observed among IBV serotypes [15].
IBVs have been isolated and identified since 1982 in China. Various live-attenuated and inactivated vaccines derived from Massachusetts (Mass) serotype strains such as H120 and H52 have been widely and extensively used in chicken farms to prevent IB disease [16, 17]. However, the efficacy of current vaccine is poor as new serotypes or antigenic variant strains of IBV continue to emerge in China, and IB disease breaks out frequently even in vaccinated flocks. Since the QX strain was detected in Shandong province, China in 1990s, the QX-like genotype IBV mainly causing nephritis has become predominant in China [18–21].
This study was a part of a long-term surveillance program that aimed at identifying IBV strains isolated in commercial chicken farms in southern China. During 2011–2012, 62 IBV strains were isolated from flocks with clinical symptoms. The genetic analysis of the newly isolates was conducted by sequencing the S1 gene and performing sequence alignment, phylogenetic analysis, and recombination analysis by comparing with reference strains.
Materials and methods
Viruses
From January 2011 to December 2012, specimens (kidney, trachea, and lung) of suspected IBV-infected chicken were collected from chicken farms distributed in southern China. Typical clinical signs of diseased birds included respiratory, nephritis, and egg drop symptoms. The specimens were frozen and thawed three times, treated with phosphate-buffered saline (PBS) containing 200 U/ml penicillin and 200 µg/ml streptomycin and then centrifuged at 7,000×g for 5 min. After keeping in 4 °C for 3 h, the viruses were propagated by blind passaging three times. In each blind passage, 9-day-old embryos of specific pathogen-free (SPF) chickens were inoculated with 0.2 ml of the supernatant of each isolate via the allantoic cavity. The allantoic fluids were harvested after incubation at 37 °C for 48 h post-inoculation. The presence of IBV was identified and verified by reverse transcription-polymerase chain reaction (RT-PCR) for the N gene.
S1 gene primers
A pair of primers used for amplifying the entire S1 gene (sense primer: 5′-AAG ACT GAA CAA AAG ACC GAC T-3′ and anti-sense primer: 5′-CAA AAC CTG CCA TAA CTA ACAT A-3′) was designed by Primer Premier 5.0 software based on alignment of GenBank sequences of several published IBV strains from China. The amplified segment was anticipated to be about 1760 bp, including the entire S1 gene and the protease cleavage motif. Primers were synthesized by AuGCT DNA-SYN Biotechnology Co., Ltd (Beijing, China).
RNA extraction and RT-PCR amplification
Viral RNA was extracted using the AxyPrep™ Body Fluid Viral DNA/RNA Miniprep Kit (Axygen, Hangzhou, China) according to the manufacturer’s instructions. The S1 gene was amplified using the PrimeScript One Step RT-PCR Kit Ver. 2 (Takara, Dalian, China) in a 50 μl reaction volume containing 2 μl of PrimeScript One Step Enzyme Mix, 25 μl of 2× One Step Buffer, 3 μl of extracted viral RNA, 2 μl of the specific primer pair, and 18 μl of RNase-free dH2O. Reverse transcription was performed with one cycle of 50 °C for 30 min and 94 °C for 5 min. PCR was followed by 30 cycles of denaturation at 94 °C for 35 s, annealing at 51 °C for 35 s, and extension at 72 °C for 90 s, followed by a final 10 min extension step at 72 °C. The products were analyzed by electrophoresis on a 1.0 % agarose gel and then observed using an ultraviolet transilluminator.
DNA cloning
PCR products of each RT-PCR were purified using the AxyPrep™ DNA Gel Extraction Kit (Axygen), ligated to the TA cloning vector pMD19-T (TaKaRa) and transformed into DH5a E. coli competent cells. Cells carrying the recombinant plasmid were selected on Luria-Bertani (LB) agar plates containing ampicillin (100 μg/ml). Positive clones were screened by PCR with the same conditions as those for the above-mentioned PCR amplification and then sequenced by Shanghai Sang-gong Biological Engineering Technology & Services Co., Ltd (Shanghai, China).
Sequence analysis of the S1 gene
The obtained nucleotide sequences and deduced amino acid sequences of S1 genes of IBV isolates were aligned using the Editseq program in the Lasergene package (DNASTAR, Madison, WI, USA) and analyzed for homology with those of 40 other reference IBV strains using the MegAlign program in the same package. Phylogenetic analysis of the nucleotide sequences and deduced amino acid sequences of the S1 gene was performed with the neighbor-joining method using MEGA version 5.0. The bootstrap values were determined from 1,000 replicates of the original data.
Recombination analysis of S1 gene
Each putative recombinant sequence and its parental strains were identified using RDP program version 4.36 with the recombination detection methods (RDP, GENECONV, BootScan, MaxChi, Chimaera, and SiScan) with default setting [22]. The potential recombination events were further verified by SimPlot version 3.5.1 [23]. The nucleotide identity was performed by the Kimura (2-parameter) method with a transition–transversion ratio of 2, and the window width and step size were 200 and 20 bp, respectively. BootScan analysis was also carried out employing a subprogram embedded in SimPlot, using signals of 70 % or more of the observed permuted trees to indicate potential recombination events [24].
IBV strains published in Genbank
Forty representative IBV strains published in Genbank were selected for the phylogenetic and alignment analysis, including Ma5, H120, H52, W93, M41, Beaudette, Ark99, Gray, Holte, 7/93, 4/91, LX4, QXIBV, A2, CQ04-1, DY05, HN08, IBVSX4, LZ05, LZ07, SAIBK, TC07-2, TA03, CK/CH/GD/KP10, CK/CH/GD/NC10, CK/CH/GX/NN09, CK/CH/LJS/08II, CK/CH/LSD/08-12, CK/CH/SC/ZJ10-1, T07/02, 3468/07, CK/CH/GD/HY09, CK/CH/GX/YL09-2, CK/CH/GD/LZ09, CK/CH/FJ/PT10, CK/CH/JX/JA09-1, 3263/04, 3071/03, TW2575/98, and CK/CH/HuN/NX09 (Table 2).
Results
Virus isolation
During the period of 2011–2012, sixty-two IBV strains were isolated from infected chicken farms distributed in provinces of southern China, including Guangdong, Guangxi, Fujian, Jiangsu, Anhui, Zhejiang, Sichuan, Yunnan, Hunan, and Hubei provinces. All the isolated strains were identified by RT-PCR for the N gene. The main features of each IBV strain are presented in Table 1. According to the analysis, 66.1 % of the new strains (41/62) were isolated in the winter and spring, which are cold and wet seasons in southern China. The age of the infected chickens, including 57 flocks of broilers and 5 flocks of layer hens, ranged from 9 to 285 days. Of the 62 new strains isolated, 37 (59.6 %) were from chickens between 10 to 30 days of age; 41 (66.1 %) were from chickens showing typical nephritis symptoms such as swollen specked kidney, distended ureter filled with uric acid, and severe dehydration and weight loss; 16 (25.8 %) were from chickens showing typical respiratory clinical signs including gasping, coughing, sneezing, tracheal rale, and nasal discharge; and 5 (8.1 %) caused a decline in egg production and quality of layer hens.
Homologies of S1 nucleotides and deduced acid sequences among isolates and reference strains
The S1 gene sequences of the 62 isolated IBV strains were submitted to Genbank (accession numbers are shown in Table 1). Homology analysis of the 62 isolated strains revealed nucleotide and deduced amino acid sequence similarities ranging from 65.9 % (strains CK/CH/SC/MS12-1 and CK/CH/GX/NN11-4) to 99.9 % (strains CK/CH/SC/ZJ11 and CK/CH/SC/MS11-1) and 58.1 % (strains CK/CH/GX/LC11-3 and CK/CH/GD/CG11) to 100 % (strains CK/CH/SC/ZJ11 and CK/CH/SC/MS11-1), respectively. When comparing with the 40 published reference strains (Table 2), the similarities of the nucleotide and deduced amino acid sequences among all of the 102 strains ranged from 63.5 to 99.9 and 57.9 to 100 %, respectively. These results indicated low homology and high variation of S1 among the isolated and reference strains.
Phylogenetic clustering of 62 isolated IBV strains into six groups
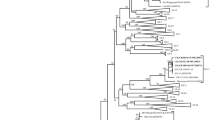
In order to determine the genetic relationships among the IBV strains, a phylogenetic tree was constructed using the nucleotide sequences of the S1 gene of the 62 isolates and 40 reference IBV strains. As shown in Fig. 1, all strains clustered into eight distinct genetic groups. All of the Chinese strains and most reference strains clustered into six groups (I–VI). Most of the current vaccine strains (H120, H52, W93, and Ma5) formed the group MASS, while the group Gray mainly consisted of classical American strains, such as Ark99, Holte, and Gray. Strains in groups I–VI showed nucleotide sequence similarities of 86.5–99.8, 85.1–99.3, 97.5–99.7, 88.2–99.7, 86.5–99.9, and 98.3–99.4 %, respectively.

Phylogenetic tree of 62 isolates and 40 reference strains (filled triangle) for S1 genes of IBVs (starting at the AUG translation initiation codon and ending at the cleavage recognition motifs). The phylogenetic tree was constructed using the MEGA version 5.0 by the neighbor-joining method with No. of differences model and setting bootstrap 1,000 replicates
Group I, which consisted of 8 reference strains and 32 new strains isolated from all provinces in southern China, was designated as the QX-like type for inclusion of the Chinese QXIBV strain. Group II, which comprised 5 reference strains and 7 new strains isolated from the provinces of Guangdong, Guangxi, Yunnan, and Sichuan, was designated as the 4/91-like type for the inclusion of 4/91 strain. Three reference strains and six new strains were included in group III. All of the new strains in this group were isolated from the provinces of Guangxi and Sichuan. Five reference strains and three new strains, which were all isolated from Guangxi province, formed group IV. Group V, also called the TW type for the inclusion of Taiwanese reference strains, had low similarity with strains from mainland China. Group V was separated into two subgroups: TW I and TW II. This group included 5 Taiwanese reference strains (TW2575/98, 3468/07, 3071/03, 3263/04, and T07/02), 2 Chinese reference strains (CK/CH/HuN/NX09 and CK/CH/SC/ZJ10-1), and 11 new strains isolated from the provinces of Sichuan, Fujian, Guangxi, Zhejiang, and Yunnan. Group VI comprised three reference strains and three new strains isolated from the provinces of Guangxi and Guangdong. The results illustrate a highly complex pattern of IBV epidemiology in China. The vaccine strains (group MASS) and foreign strains (group Gray) were evolutionarily distant from the Chinese IBV strains according to the branching of the phylogenetic tree.
Mutational analysis
Alignments of the S1 gene nucleotide and deduced amino acid sequences of all 62 isolated strains showed that they contain 1611, 1614, 1617, 1620, 1623, 1626, 1629, 1632, and 1635 nucleotides (position from ATG start site to the cleavage recognition site), corresponding to 537, 538, 539, 540, 541, 542, 543, 544, and 545 amino acids (Table 1). This analysis indicated that the S1 genes of the newly isolated strains contain mutations, insertions, and deletions, resulting in different lengths of nucleotide and amino acid sequences.
The alignment analysis of the deduced amino acid sequences of S1 from all 62 isolated strains with reference strain H120 revealed three hypervariable regions (HVRs) located between amino acid residues 60–88, 115–140, and 275–292 (numbered according to H120 reference S1 sequence). The featured deletions, insertions, and mutations are summarized in Fig. 2.

Sequence alignment of S1 amino acid sequences of isolated strains with the H120 reference strain. A dot indicates an amino acid identical to that of the H120 strain. A dash indicates an amino acid deletion in comparison with the H120 strain
The precursor protein of the S glycoprotein is cleaved into S1 and S2 subunits by the cellular protease during viral maturation [25]. Cleavage recognition motifs of the 62 isolates and 40 reference IBV strains are listed in Tables 1 and 2, respectively. In this study, 6 cleavage recognition motifs of the S protein were found among the 62 isolated strains and compared with the 8 cleavage recognition motifs among the 40 reference strains. Five of the cleavage site motifs (HRRRR, RRFRR, RRLRR, HRRKR, and RRSRR) are shared by both the new isolates and reference strains. The common cleavage recognition sites of the S protein were HRRRR (32/62), RRF(S/L) RR (26/62), and HRRKR (3/62) in the isolated strains (Table 2). The exception was CK/CH/SC/MS12-1 (RRIRR), containing the amino acid I in the cleavage sites, which was quite different from those of the other isolates and reference strains.
S1 gene recombination
Recombination events of isolated IBV strains were detected using the RDP software. In this study, recombination events in group I subgroup (Fig. 1) were observed. The result is shown in Table 3. The S1 genes of the two group I subgroup strain (CK/CH/SC/DY12-2 and CK/CH/ZJ/QZ12-2) both share high similarity (99.8 and 100 %) with minor parental strain TA03 (group II) between break point 889 and 1458 nts, and the rest part of S1 genes shared a high sequence identity (both 99.3 %) with the major parental strain CK/CH/GD/LZ09 (group I). The two recombination events were detected by all the 6 methods (RDP, GENECONV, BootScan, MaxChi, Chimaera, and SiScan) and the p value calculated by RDP method were both 2.969 × 10−50. The two recombination events were further verified by Simplot software. Similarity analysis and BootScan analysis on the putative recombinant strains and their parental sequence were conducted. Strains were considered as recombinants if any crossover event appeared between two putative parental strains. The result is shown in Fig. 3. The obvious recombination signals were found in Similarity and BootScan analysis of CK/CH/SC/DY12-2 and CK/CH/ZJ/QZ12-2. The similarity and break point agreed with the RDP software.

Similarity analysis (a) and BootSan analysis (b) on the putative recombinant CK/CH/ZJ/QZ12-2 and CK/CH/SC/DY12-2. Reference strain CK/CH/GD/LZ09 (red) and TA09 (blue) were used as putative parental strains. M41 (gray) was used as an outlier sequence. The query sequence is indicated on the upper part of the figure. The y-axis gives the percentage of identity within a sliding window 200 bp wide centered on the position plotted, with a step size between plots of 20 bp
Discussion
This study is part of a long-term surveillance program with the purpose of researching the epidemiology of IBV and identifying IBV strains emerging in commercial chicken farms in southern China. Despite the wide use of attenuated live vaccines such as H120, H52, Ma5, and 4/91, IB disease still caused persistent infection and frequent outbreak in commercial chicken farms [26, 27].
In this study, 62 strains of IBV were isolated from diseased chicken flocks in southern China during the period between 2011 and 2012. As shown in Table 1, the isolation rate of IBV was closely related to the pattern of IB outbreak. Generally, IB disease can break out in any season and infect chickens of any age, but in southern China IB disease appears to have infected mostly chickens less than 30 days old and to break out in the winter and spring seasons when it was cold and wet. The classical IBV strains mainly affect respiration in chickens of all ages and cause a drop in egg production in layer hens. However, in addition to respiratory complications, nephritis gradually became the major clinical sign of IBV infection from the late 1990s in China, and proventriculitis was also reported [28, 29]. In this study, the recorded clinical signs showed that 66.1 % (41/62) of the identified isolates exhibited typical nephritis. These findings support accumulating evidence indicating that nephropathogenic IBV strains have become prevalent in China.
The phylogenetic analysis indicated a complicated pattern of epidemiology of IBVs in southern China, with six distinct genetic groups co-circulating in the field and multiple strains which may be responsible for the constant IB outbreaks. All of the isolates and reference strains clustered into 8 groups: group MASS, group Gray, and 6 Chinese groups. The Chinese groups were evolutionarily distant from the vaccine and foreign groups. It suggested that the evolutionary distant may contribute to the inefficiency of vaccine and the vaccine strains need to be updated. Among the 6 Chinese genogroups, group I, group V, and group VI were of note.
Group I (QX-like type) is the predominant genogroup in China and accounted for 50–60 % of the isolated stains in recent years [18–21]. In this study, 51.6 % (32/62) of the isolated strains belonged to group I and were distributed in all provinces of southern China. QXIBV, first isolated in Shandong province, China in 1996, was associated with proventriculitis, while the QX-like strains prevalent in China mainly caused typical nephritis and false layers. QX-like IBV strains also were reported to have become the primary genotype in other areas of Asia and some European countries [30]. Considering that Group I has become the predominant genotype, it is necessary to develop QX-like vaccine strains to control the IBV infection in China.
Group V (TW type) was separated into 2 subgroups: TW I and TW II. According to our long-term surveillance, IBV strain in this group was seldom isolated and mainly isolated in Sichuan province before 2010. However, in the period of present study, the isolation rate of this group strains soared to 17.7 % (11/62) and isolated in most provinces of southern China. Whether group V strains have become the main genogroup in some areas needs further study. It was reported that TW-type IBV strains were different from strains isolated from mainland China and they had different origins [31]. In this study, group V shared nucleotide sequence identities of only 65.1–83 % with other groups in S1 gene. IBV strains in this group were isolated in most provinces of southern China, such as Sichuan, Guangdong, Guangxi, Fujian, Hunan, Zhejiang, and Yunnan provinces. It suggested that TW-type strains have widely involved in the genetic evolution of IBV in China. The island of Taiwan is geographically separated from mainland China by the Taiwan Strait. Considering that there is no live poultry trade and no TW-type vaccine inflow into mainland China, migrating birds were speculated to provide the genetic sources of TW-type variants [32].
Group VI comprised three isolated strains and three reference strains (TC07-2, CK/CH/GD/NC10, and CK/CH/GD/KP10). This group shared nucleotide sequence identities of only 63.5–67.4 % with other groups in the S1 gene and showed a distant relationship with other strains. Since the reference strain TC07-2 was first isolated in Guangdong province in 2007, a small number of strains in this group have been only isolated in Guangdong and Guangxi provinces in southern China and also reportedly isolated in Japan in 2009 and Korea in 2010 [33, 34].
The S1 glycoprotein determined genetic diversity, phenotype change, and serotypic evolution of IBVs. In the present study, nine different lengths of S1 nucleotide or deduced amino acid sequences were found, and the predominant length (nt/aa) was 1620/540. Alignments of S1 genes from the 62 isolates along with 40 reference strains showed a wide range of homologies among them. Many reasons may account for those sequence differences, the most important is that IBV is a non-segmented single-strand positive-sense RNA virus, and its error-prone RNA polymerase can easily generate nucleotide insertions, deletions, and point mutations in the S1 gene. This trait brings about not only variations of viral gene and protein sequences, but also potential changes in virulence and tissue tropism. Alignment of the S1 amino acid sequences revealed that there were three HVRs located at 60–88, 115–140, and 275–292 (numbered based on the S1 sequence of the H120) among the 62 isolates, similar to the previous studies [35–37]. It was obvious that the HVR was different from other genogroup strains. Actually, the divisions of genotypes were partly according to the HVRs [38, 39]. It was also reported that the serotype differences of IBVs generally correlated with variations in HVR of S1 protein [40, 41]. However, the significance of HVRs in IBV pathogenicity remains unclear.
The cleavage recognition motif of S1 gene is reportedly irrelevant to the viral pathogenicity and tissue tropism [42], although it shows continual evolution in IBV strains in the field. In this study, six types of S1 protein cleavage recognition motifs were found among the isolates, none of which were newly emerged. The predominant S1 protein cleavage recognition motifs were HRRRR (33/62) and RRF(S/L) RR (26/62). The motif RRIRR was first reported in 2011 [43], and it is not common in isolates from the field. The motif HRRRR is consistently present in Chinese IBV strains but not commonly observed in viruses from other countries [44], showing the continuity of evolution and co-circulation of many viruses with putative separate origins in specific geographic regions [45].
Recombination is another important mechanism of IBV evolution to generate variants in the field [46]. The unique discontinuous transcription system and the viral polymerase “jumping” possibly contribute to the high RNA recombination frequency in IBVs [47]. The recombination event is thought to occur by switching of the polymerase from one template to another during the genomic synthesis when the host is infected by two or more strains of IBV [17]. The S1 gene recombination reportedly took place between the Chinese QXIBV and classical IBV strains in countries around China, such Thailand and Korea, producing new variant strains and subgroups [23, 33, 48]. In this study, two isolated strains were detected as recombinant variants, and the recombination events between group I (QX-like) strain and group II (4/91-like) strain formed a new subgroup of group I. The putative major parental strain CK/CH/GD/LZ09, isolated in Guangdong province in 2009, was a typical QX-like strain-caused nephritis. The putative minor parental strain TA03 was reported as a real 4/91-like variant strain which was rarely isolated in China and the pathogenicity and virulence were verified by artificial infection [18]. The recombination analysis suggested that 4/91-like strains, as important genetic donor, have involved in the genetic recombination of IBV in China. The results also indicate that genetic recombination contributes to the emergence of new variants and sub-genogroups in southern China. Considering that the S1 gene is only a small part of IBV genome, the change of virulence, tissue tropism, and pathogenicity of the two strains caused by recombination need to be further studied.
In conclusion, the present study suggests that six genogroups of IBV strains are co-circulating in commercial chicken farms in southern China, with most of the isolates belonging to group I. It is worthwhile to note that the isolation of group V strains had been increasing during the two-year observation period. The vaccine strains were evolutionarily distant from isolates and should be updated. The emergence of new strains and sub-genogroups can be attributed to sequence changes, such as mutations, insertions, deletions, and recombination events, which promote viral evolution. This work is part of our ongoing long-term surveillance program and highlights the importance of continuing to monitor the new IBV strains.
References
L. Yu, Y. Jiang, S. Low, Z. Wang, S.J. Nam, W. Liu, J. Kwangac, Avian Dis. 45, 416–424 (2001)
D. Cavanagh, P.J. Davis, J.K. Cook, D. Li, A. Kant, G. Koch, Avian Pathol. 21, 33–43 (1992)
D. Cavanagh, J. Gen. Virol. 64(Pt 12), 2577–2583 (1983)
D. Cavanagh, P.J. Davis, J.H. Darbyshire, R.W. Peters, J. Gen. Virol. 67(Pt 7), 1435–1442 (1986)
D. Cavanagh, P.J. Davis, J. Gen. Virol. 67(Pt 7), 1443–1448 (1986)
G. Koch, L. Hartog, A. Kant, D.J. van Roozelaar, J. Gen. Virol. 71(Pt 9), 1929–1935 (1990)
H.M. Kwon, M.W. Jackwood, T.P. Brown, D.A. Hilt, Avian Dis. 37, 149–156 (1993)
B.F. Kingham, C.J. Keeler, W.A. Nix, B.S. Ladman, J.J. Gelb, Avian Dis. 44, 325–335 (2000)
D.F. Stern, B.M. Sefton, J. Virol. 44, 794–803 (1982)
D. Cavanagh, P.J. Davis, J.H. Darbyshire, R.W. Peters, J. Gen. Virol. 67(Pt 7), 1435–1442 (1986)
S. Duffy, L.A. Shackelton, E.C. Holmes, Nat. Rev. Genet. 9, 267–276 (2008)
N. Tromas, M.P. Zwart, M. Poulain, S.F. Elena, J. Gen. Virol. 95, 724–732 (2014)
P.C. Woo, S.K. Lau, K.Y. Yuen, Curr. Opin. Infect. Dis. 19, 401–407 (2006)
F. Cong, X. Liu, Z. Han, Y. Shao, X. Kong, S. Liu, BMC Genom. 14, 743 (2013)
C.J. Keeler, K.L. Reed, W.A. Nix, J.J. Gelb, Avian Dis. 42, 275–284 (1998)
S.W. Liu, Q.X. Zhang, J.D. Chen, Z.X. Han, X. Liu, L. Feng, Y.H. Shao, J.G. Rong, X.G. Kong, G.Z. Tong, Arch. Virol. 151, 1133–1148 (2006)
S. Liu, X. Zhang, Y. Wang, C. Li, Z. Han, Y. Shao, H. Li, X. Kong, Intervirology 52, 223–234 (2009)
Z. Han, C. Sun, B. Yan, X. Zhang, Y. Wang, C. Li, Q. Zhang, Y. Ma, Y. Shao, Q. Liu, X. Kong, S. Liu, Infect. Genet. Evol. 11, 190–200 (2011)
L. Li, C. Xue, F. Chen, J. Qin, Q. Xie, Y. Bi, Y. Cao, Vet. Microbiol. 143, 145–154 (2010)
J. Ji, J. Xie, F. Chen, D. Shu, K. Zuo, C. Xue, J. Qin, H. Li, Y. Bi, J. Ma, Q. Xie, Virol. J. 8, 184 (2011)
H. Luo, J. Qin, F. Chen, Q. Xie, Y. Bi, Y. Cao, C. Xue, Virus Genes 44, 19–23 (2012)
L. Heath, E. van der Walt, A. Varsani, D.P. Martin, J. Virol. 80, 11827–11832 (2006)
T.H. Lim, H.J. Lee, D.H. Lee, Y.N. Lee, J.K. Park, H.N. Youn, M.S. Kim, J.B. Lee, S.Y. Park, I.S. Choi, C.S. Song, Infect. Genet. Evol. 11, 678–685 (2011)
T. Pohuang, N. Chansiripornchai, A. Tawatsin, J. Sasipreeyajan, Virus Genes 43, 254–260 (2011)
D. Cavanagh, P.J. Davis, J.K. Cook, D. Li, A. Kant, G. Koch, Avian Pathol. 21, 33–43 (1992)
C. Xu, J. Zhao, X. Hu, G. Zhang, Vet. Microbiol. 122, 61–71 (2007)
G.X. Bing, X. Liu, J. Pu, Q.F. Liu, Q.M. Wu, J.H. Liu, Virus Genes 35, 333–337 (2007)
S. Liu, X. Kong, Avian Pathol. 33, 321–327 (2004)
L. Yu, Y. Jiang, S. Low, Z. Wang, S.J. Nam, W. Liu, J. Kwangac, Avian Dis. 45, 416–424 (2001)
H.J. Geerligs, G.J. Boelm, C.A.M. Meinders, B.G.E. Stuurman, J. Symons, J. Tarres-Call, T. Bru, R. Vila, M. Mombarg, K. Karaca, W. Wijmenga, M. Kumar, Avian Pathol. 40, 93–102 (2011)
S.W. Liu, Q.X. Zhang, J.D. Chen, Z.X. Han, X. Liu, L. Feng, Y.H. Shao, J.G. Rong, X.G. Kong, G.Z. Tong, Arch. Virol. 151, 1133–1148 (2006)
H.W. Chen, Y.P. Huang, C.H. Wang, Virus Res. 140, 121–129 (2009)
T.H. Lim, M.S. Kim, J.H. Jang, D.H. Lee, J.K. Park, H.N. Youn, J.B. Lee, S.Y. Park, I.S. Choi, C.S. Song, Poult. Sci. 91, 89–94 (2012)
M. Mase, N. Kawanishi, Y. Ootani, K. Murayama, A. Karino, T. Inoue, J. Kawakami, J. Vet. Med. Sci. 72, 1265–1268 (2010)
D. Cavanagh, P.J. Davis, A.P. Mockett, Virus Res. 11, 141–150 (1988)
S.W. Liu, Q.X. Zhang, J.D. Chen, Z.X. Han, X. Liu, L. Feng, Y.H. Shao, J.G. Rong, X.G. Kong, G.Z. Tong, Arch. Virol. 151, 1133–1148 (2006)
K.M. Moore, M.W. Jackwood, D.A. Hilt, Arch. Virol. 142, 2249–2256 (1997)
J.K. Cook, M. Jackwood, R.C. Jones, Avian Pathol. 41, 239–250 (2012)
D. Cavanagh, Vet. Res. 38, 281–297 (2007)
S.A. Callison, M.W. Jackwood, D.A. Hilt, Avian Dis. 45, 492–499 (2001)
D. Cavanagh, P.J. Davis, J. Gen. Virol. 69(Pt 3), 621–629 (1988)
M.W. Jackwood, D.A. Hilt, S.A. Callison, C.W. Lee, H. Plaza, E. Wade, Avian Dis. 45, 366–372 (2001)
F. Yan, Y. Zhao, W. Yue, J. Yao, L. Lihua, W. Ji, X. Li, F. Liu, Q. Wu, Avian Dis. 55, 451–458 (2011)
G.X. Bing, X. Liu, J. Pu, Q.F. Liu, Q.M. Wu, J.H. Liu, Virus Genes 35, 333–337 (2007)
M.W. Jackwood, D.A. Hilt, S.A. Callison, C.W. Lee, H. Plaza, E. Wade, Avian Dis. 45, 366–372 (2001)
C.W. Lee, M.W. Jackwood, Virus Res. 80, 33–39 (2001)
N.L. Zou, F.F. Zhao, Y.P. Wang, P. Liu, S.J. Cao, X.T. Wen, Y. Huang, Virus Genes 41, 202–209 (2010)
T. Pohuang, N. Chansiripornchai, A. Tawatsin, J. Sasipreeyajan, Virus Genes 43, 254–260 (2011)
Acknowledgments
This study was supported by the Natural Science Foundation of Guangdong Province (Grant No. S2013030013313) and the Technology Planning Project of Guangdong Province of China (Grant Nos. 2012B020306002 and 2012B091100078).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Feng, K., Xue, Y., Wang, F. et al. Analysis of S1 gene of avian infectious bronchitis virus isolated in southern China during 2011–2012. Virus Genes 49, 292–303 (2014). https://doi.org/10.1007/s11262-014-1097-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-014-1097-1