
Abstract
The complete genome sequence of the KGH strain of the first human rabies virus, which was isolated from a skin biopsy of a patient with rabies, whose symptoms developed due to bites from a raccoon dog in 2001. The size of the KGH strain genome was determined to be 11,928 nucleotides (nt) with a leader sequence of 58 nt, nucleoprotein gene of 1,353 nt, phosphoprotein gene of 894 nt, matrix protein gene of 609 nt, glycoprotein gene of 1,575 nt, RNA-dependent RNA polymerase gene of 6,384 nt, and trailer region of 69 nt. Sequence similarity was compared with 39 fully sequenced rabies virus genomes currently available, and the result showed 70.6–91.6 % at the nucleotide level, and 82.8–97.9 % at the amino acid level. The deduced amino acids in the viral protein were compared with those of other rabies viruses, and various functional regions were investigated. As a result, we found that the KGH strain only had a unique amino acid substitution that was identified to be associated either with host immune response and pathogenicity in the N protein, or with a related region regulating STAT1 in the P protein, and related to pathogenicity in G protein. Based on phylogenetic analyses using the complete genome of 39 rabies viruses, the KGH strain was determined to be closely related with the NNV-RAB-H strain and transplant rabies virus serotype 1, which are Indian isolates, and was confirmed to belong to the Arctic-like 2 clade. The KGH strain was most closely related to the SKRRD0204HC and SKRRD0205HC strain when compared with Korean animal isolates, which was separated around the same time and place, and belonged to the Gangwon III subgroup.
Similar content being viewed by others


Introduction
Rabies is a major threatening zoonotic disease of humans and animals, which has been reported since 2300 BC [1, 2]. In 1802, rabies was reported as a communicable disease that could be transferred from rabid to healthy dogs via the saliva [3], and was verified to be transmitted from one animal species to another [4, 5]. Although rabies virus vaccines have been developed, human rabies is often misunderstood as a neglected infectious disease, and hence, results in some 55,000 deaths annually, mainly in Asia and Africa [6].
The rabies virus is a member of the family Rhabdoviridae, genus Lyssavirus. Lyssavirus has seven genotypes. The most widespread classical rabies viruses belong to genotype 1, and virus isolates derived from Africa, Asia, and European bat species are genotypes 2–7 [7, 8]. The rabies virus genome is a negative-sense, single-stranded RNA, and the RNA genome is tightly encapsidated by nucleoprotein [6]. The genome is approximately 12 kb, and is encoded with five structural proteins, including nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and RNA-dependent RNA polymerase (L).
The N protein forms the ribonucleoprotein complex together with the P and L proteins and viral genomic RNA to regulate RNA transcription and replication, evade activity of RIG-I-mediated signaling which is a host innate immune response, and affect pathogenicity [9]. The G protein mediates easy entry of the virus into cells, affects viral spread and uptake speed, and plays an important role in neurovirulence and selection of antigenic sites [6, 10]. Phylogenetic analyses have focused on the use of partial sequences of the N or G genes in other rabies virus strains. The rabies virus N gene is mostly used for evolutionary studies, because it is well conserved. The G gene is mostly used for epidemiological and evolutionary studies, because it changes amino acids at antigenic sites to escape from the host immune system [11, 12]. As the G–L non-coding region is not affected by positive selection, it is used for phylogenetic analyses [12]. Host-specific features can be confirmed through determining the complete genome sequence, and very subtle changes in unknown regions of the viral genome can be identified [13]. These methods of analysis contribute to understanding the origin and transmission pattern of the disease, and provide an efficient strategy for regulating rabies based on molecular epidemiological data [14].
Rabies was reported for the first time in Korea in 1907. Since then, an average of 500 cases per year were reported until 1984, when the incidence dropped to 30 cases in 1985 following the widespread vaccination of dogs. No incidence of rabies was reported between 1985 and 1992, but animal rabies cases were reported during the period of 1993–2004 [15]. No human rabies case was reported between 1985 and 1998, but a single case in 1999, and five further cases up until 2004 were reported. Partial sequences of the N, G, P, and G–L non-coding regions have been analyzed, but the complete genome sequence analysis is reported for the first time here [15, 16]. Through maximum-parsimony analysis, strains of rabies viruses, which are prevalent in Asia, were divided into five distinct clusters, including Asian 1, Asian 2, Asian 3, Arctic-related, and Cosmopolitan clusters [17]. The Arctic-like lineage is composed of the Arctic-like 1 and Arctic-like 2 clades [18]. According to a recent classification, Korean isolates belong to the Arctic-like 2, and consist of the four sub-groups of Gangwon I, Gangwon II, Gangwon III, and Gyeonggi [16, 18].
In the present study, we have determined the complete genome sequence of the KGH strain, which was the first human rabies virus isolated from a skin biopsy of a Korean patient with rabies in 2001. The KGH strain was compared to 39 fully sequenced rabies virus strains at the molecular level. A complete genome sequence analysis is a basis for understanding the molecular characteristics and genetic diversity of rabies viruses prevalent in Korea. In particular, a protein substitution associated with the host immune response, pathogenicity, and signal transducers and activators of transcription (STAT)1 regulation was observed in the KGH strain. Our results will aid the understanding of the immune response of rabies viruses prevalent in Korea, as well as the identification of their relationship with pathogenicity.
Materials and methods
Virus isolation
The KGH rabies virus strain was isolated from the hair follicles of a 68-year old patient who was bitten by a rabid raccoon dog in Hwacheon-gun, Gangwon-do in 2001 and that had an 11-week incubation period without PEP treatment. The patient was observed for symptoms of hyperesthesia, headache, nausea, photophobia, and hydrophobia. This rabies virus strain was isolated from the brains of suckling mice that were intracerebrally injected with homogenized hair follicles of the patient. The brain tissues were 50 % homogenated in a tube with PBS and ceramic beads.
Reverse transcription and amplification
Viral RNA was extracted from 200 μl of a 10 % brain homogenate from the infected mice using Trizol LS reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s recommendations. Extracted viral RNA was resuspended in 30 μl of diethylpyrocarbonate-treated water (Invitrogen, USA). Superscript III (Invitrogen, USA) was used for the first-strand cDNA synthesis according to the manufacturer’s instructions. Three synthesized cDNA fragments (RV24F, RV3866F, and RV6325F) were amplified by polymerase chain reaction (PCR) using Pyrobest DNA polymerase (Takara Bio Inc., Shiga, Japan), and the primers are listed in Table 1. PCR primer sets for amplifying the RV24F amplicon (nt 1–4178) were RV24F and RV4178R, those for amplifying the RV3866F amplicon (nt 4068–8003) were RV4068F and RV8003R, and those for amplifying the RV6325F amplicon (nt 7623–11928) were RV7623F and RV11928R. The PCR parameters included denaturation at 94 °C for 30 s, annealing at 52° C for 30 s, extension at 72° C for 5 min, and a final extension at 72° C for 10 min. After electrophoresis with 1 % agarose gels, all PCR products were purified using QIAquick Gel Extraction kit (Qiagen, Hilden, Germany). Direct sequencing was conducted twice in both directions.
5′-Random amplification of cDNA ends (RACE) PCR and RLM-3′-RACE
A standard 5′-RACE kit (Invitrogen, USA) was used to determine the 5′-terminal ends of the genomic RNA. The reaction was performed according to the manufacturer’s instructions. For the first-strand cDNA synthesis, the RVLF gene specific primer with Superscript III RT (Invitrogen, USA) was used with 8 μl of viral RNA. The synthesized cDNA was purified in a 30 μl final volume using a S.N.A.P. column purification kit (Invitrogen, USA). A 10 μl aliquot of purified cDNA was used for the TdT-tailing reaction. For the first-round PCR, 5 μl of dC-tailed cDNA and an anti-sense abridged anchor primer included in the 5′RACE kit were used with the RVLF positive sense primer. The RVLFN internal positive sense primer and abridged anchor primer were used for the second-round PCR with a pair of primers. All PCR reactions were amplified with Pyrobest DNA polymerase with the following thermocycling parameters: 25 cycles (for the first-round PCR) and 30 cycles (for the second-round PCR) of 30 s at 94° C, 30 s at 60° C, 30 s at 72° C, and a final extension of 10 min at 72° C. The PCR products were inserted into the pJET vector (Fermentas, Burlington, ONT, Canada) and cloned in ECOS E. coli DH10B competent cells (Yeastern, Taipei, Taiwan). We confirmed that 50 independent clones were randomly selected for sequencing.
Genomic RNA was identified by 3′-RLM-RACE to determine the 3′-end. Viral RNA and the 5′-phosphorylated oligonucleotide 3′-P end were ligated using T4 RNA ligase (New England Biolabs, Ipswich, MA, USA) at 16° C for 2 h. RNA-3′-P end ligated RNA was recovered with TRIzol LS reagent and was used for cDNA synthesis with 3′End Rev, which is a complementary oligonucleotide. PCR was amplified with Pyrobest DNA polymerase using a pair of primers, 3′End Rev and the R470 negative-sense primer. The thermocycling parameters were 30 cycles of 30 s at 94° C, 30 s at 55° C, 50 s at 72° C, and a final extension for 10 min at 72° C. The PCR products were inserted into the pJET vector and cloned in E. coli DH10B competent cells. We confirmed that 80 independent clones were randomly selected for sequencing.
Phylogenetic and genetic distance analyses
As a full genome sequence dataset, 39 full-length nucleotide sequences of other rabies virus strains listed on available in Genbank, and 23 glycoprotein gene sequence of rabies virus strains separated in Korea, were used as shown in Table 2. Full sequences were assembled using the Seqman program of DNAstar, version 5. Multiple sequence alignments were performed using the Megalign tool (DNAstar). Phylogenetic trees were constructed, and evolutionary distances analyses were conducted using MEGA version 5.05 using the neighbor-joining algorithm with the Kimura two-parameter model. Bootstrap values were statistically evaluated with 1,000 replicates.
Results
Comparison of nucleotide and deduced amino acids sequence of the KGH strain
To characterize the KGH strain, we determined the complete genome sequence of the first Korean rabies virus strain. The full-length RNA genome of the KGH strain was confirmed to have a length of 11.928 nt, and was composed of 58 nt genomic RNA at the 3′-terminal and 69 nt genomic RNA at the 5′-terminal. Five coding regions consisted of 1,353 nt for the N gene (450 amino acids), 894 nt for the P gene (297 amino acids), 609 nt for the M gene (202 amino acids), 1,575 nt for the G gene (524 amino acids), and 6,384 nt for the L gene (2127 amino acids). The intergenic region among the sites, at which the transcription sequence starts and stops, as well as the five genes are summarized in Table 3.
Eighty and fifty independent clones were randomly selected before analyzing their base sequences to confirm the 3′-end and 5′-end sequences of genomic RNA, respectively. As a result, 10 % of the sequences were truncated, whereas the remaining sequences showed complete alignment, and consequently, complete leader and trailer sequences were determined. Two conserved sequences in the 3′ leader and 5′ trailer were in the negative-strand RNA genomes, and they are known as cyclization motifs, which play an important role in viral RNA replication [20]. The 3′-end genomic sequence (1ACGCTTAACAAC12) and the 5′-end genomic sequence (11928ACGCTTAACAAA11917) were obtained, and the first 11 nucleotides of the 3′ and 5′ terminal regions were confirmed to be complementarily conserved.
We compared the complete KGH strain genome sequence with sequences of each of the 39 other fully sequenced rabies virus strains (Table 2) which are available from GenBank. Sequence similarity at the nucleotide level was 70.6–91.6 %, and that at the amino acid level was 82.8–97.9 % (Table 4). In particular, the KGH strain showed high similarity with NNV-RAB-H (91.6 %) and transplanted rabies virus serotype 1 (91.6 %), which are two rabies virus strains isolated in India. Less similarity was observed in RRV ON-99-2 (80.1 %) and SHBRV-10 (81.6 %) of genotype 1. When each gene of the N, P, M, G, and L proteins of the KGH strain and the 40 rabies strains were compared, the N protein showed 75–92.6 % (87.8–99.3 %), the P protein showed 63.8–91.5 % (61.6–94.6 %), the M protein showed 77.3–92.1 % (84.2–97.5 %), the G protein showed 67.3–91.2 % (72–96.8 %), and the L protein showed 74–91.9 % (86.9–98.5 %) identity ranges of nucleotide and amino acids, respectively.
Structure–function analyses
Substitutions at amino acid positions that play an important role in five viral protein-coding regions were compared using the KGH strain and the 39 fully sequenced rabies virus strains (Table 5). When the N protein of the KGH strain was compared, a unique substitution was observed only at amino acid position 394 (Tyr to Cys, described as a mutation from another rabies virus strain to the KGH strain). A total of 273 and 394 amino acids of the N protein play an important role in host innate immune response and pathogenicity, respectively [9, 21]. Furthermore, a substitution occurred in the KGH strain and in an Indian isolate (NNV-RAB-H, rabies virus serotype 1), vaccines strains (PV, RbE3-15, SRV 9, SAD B19, RV97), and RRV ON-99-2 isolates at amino acid position 135 (Ser to Phe) which is a T-cell epitope in humans and mice [22]. Antigenic sites (residues 313–337, 358–367, 374–383, and 410–413), a putative casein-type phosphorylation site (Ser 389), a residue Asn 157 and Pro 435 related to immune system evasion, and the T-helper cell epitope (residue 404–418) were conserved [9, 23–26].
Two unique substitutions were observed in the P protein (Table 5). Amino acid residues 69–177 and 268–297 are N protein binding sites [27, 28], and substitutions were observed only at amino acids 268 and 296 (Ala to Gly and Thr to Val) of the KGH strain. Five phosphorylation sites in the P protein target separate cellular kinases, and serine residue positions 63/64 code RABV protein kinase C, whereas serine positions 162/210/271 code protein kinase C [27, 29]. Among them, a substitution was uniquely observed only at residues 61/62 (Gly to Lys, Lys to Glu) in the KGH strain. Among five Met residues in the P protein, Met 1/Met 20 remains in the cytoplasm according to the CRM1-dependent nuclear export signal (NES), whereas Met 53/Met and 69/Met 83 remain in the nucleus according to the nuclear localization signal (NLS). The Met 69 residue of the KGH strain was substituted with Val from Met [30, 31]. The cytoplasmic dynein light chain (LC8)-binding motif (residues 144–148), NES (residues 49–58 and 227–232), and NLS (residues 211–214 and 260R) are conserved [31, 32].
Unique amino acid substitutions were observed in the G protein (Table 5). The amino acid residue 164–303 region plays a important role in pathogenicity [33], and substitutions were observed only at amino acids 164 (Val, Arg, Gln to Ile) of KGH and India isolated strains, and at 179 (Met to Leu) and 245 (Thr to Ala) residues in KGH strains. In the G protein, 37 and 319 residues in N-glycosylated sites and most of the antigenic sites (I, II, and III) were conserved in the KGH strain. The amino acid of G protein 333, which play an important role in determining the virulence of the rabies virus, was Arg in the KGH strain. At 194 amino acids affecting viral pathogenicity were also conserved.
Proline-rich (PPEY) motif in M protein is called as a late budding domain or L domain and it plays an important role in virion assembly and budding and has been conserved in Rhabdoviridae, Retroviridae, and Filoviridae families [34]. Amino acid residue 58 regulates RNA synthesis, and the highly hydrophobic region (residues 89–107) is related to apoptosis and pathogenesis [35, 36]. In these functional domains, the KGH strain has been conserved.
There are binding sites of six domains and various functional motifs in the L protein. Domain I includes tripeptide GHP (residues 372–374) and putative leucine zipper motif, LX6LX6LX6 (residues 237–258) [37–39]. Domain II (residues 544–563) and Domain III (residues 728–732), which are major functional domains form “polymerase module” [38, 39]. Domains IV and V showed the availability of host, a rNTP binding site or tyrosine kinase activity [37, 39, 40]. Dipeptide GG (residues 899–900) and DP (residues 916–917) are known as the first conserved motif of polymerase, domain IV has been conserved mostly in rhabdoviruses [37, 40]. Domain VI (residues 1705–1710) known as glycine-rich motif (GXGXG) is strongly conserved in all animal rhadoviruses [41]. These functional six domains have been conserved in the KGH strain.
Phylogenetic analysis
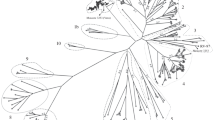
The phylogenetic relationship was analyzed using the nt sequence of the complete genome (Fig. 1). Chinese isolates and two phylogenetic clades of Arctic-like viruses were constructed as main branches. The China I and II lineages appeared with the Chinese isolates. The Arctic-like viruses were divided into two clades using the N protein nt, which were the Arctic-like-1 (AL1) clade and Arctic-like-2 (AL2) clade [18]. KRH2-04, which is a rabies virus strain isolated from Korean animals, belongs to Arctic-like-2 [16, 18, 19]. When the nt sequence of the complete KGH strain genome was compared, it was found to be closely related to the Indian isolate (NNV-RAB-H) strain, which belongs to Arctic-like-1 rather than to Japanese, Russian, or Chinese isolated strains.

Phylogenetic tree was constructed with the nucleotide sequence of the full-length genome. The tree was constructed using the neighbor-joining algorithm with the Kimura two-parameter in MEGA version 5.05. Numbers below branches are bootstrap values calculated from 1,000 replicates. West Caucasian bat virus was used as an outgroup. The KGH Korean isolate strain is marked with a red triangle (Color figure online)
The phylogenetic tree was constructed based on glycoprotein gene (Fig. 2). It was closely related to the Mongolia isolate strain, which belongs to Arctic-like-2 clade. The KGH strain was constructed with Korean rabies virus strains and is composed of four distint sub-groups; it was most closely related with Korean strains which were isolated from the same region at same time period [16, 18].

Phylogenetic tree constructed with the G gene sequence of 68 selected rabies virus strains isolated from Asia. The KGH Korean isolate strain is marked with a red triangle. The trees were used together with the Kimura two-parameter neighbor-joining algorithm in MEGA version 5.05. Numbers below branches are bootstrap values calculated from 1,000 replicates (Color figure online)
Discussion
The purpose of this study was to identify the complete genome sequence of the KGH strain, which is a rabies virus separated from the hair follicles of a Korean patient with rabies, and to compare it with 39 available fully sequenced rabies virus strains. The KGH strain had a nt length of 11,928, which is the same as NNV-RAB-H, SAD B19, and SRV9. The KGH strain was longer than H-08-1320 (11,926 nt), BR-Pfx1(11,924 nt), and Japanese isolates (11,926 nt), but was shorter than the vaccine strains of PV(11,932 nt), ERA (11,931 nt), RBE3-15(11,931nt), and RV-97(11,932nt). Diverse variations and lengths in the G–L region affect not only full genome length but also virus proliferation by regulating gene expression [42].
A unique substitution was observed in residue 394 of the KGH strain. When the avirulent Ni-CE strain was prepared through passage of the virulent Nishigahara strain (Ni) from Japan, the 273 (Phe to Leu), 394 (Tyr to His), and 395 (Phe to Leu) residues of the N protein changed [36]. Amino acids 273 and 394 of the N protein play an important role in evading RIG-I-mediated antiviral responses and have an important relationship with pathogenicity in adult mice [9, 21]. Residue 394 of the Ni and Ni-CE strains was a hydrophillic Tyr or His, whereas the KGH strain had a hydrophobic Cys. This may be due to the effects on N protein structure and residues 373–395 [22], which are T-cell epitopes, and subsequently, interferon (IFN)-β and the expression of CXCL10 [43], which is regulated by IRF-3, may be affected. Host antiviral responses may also be affected, as residue 394 of the N protein includes the P protein binding domain [36]. Rabies virus strains isolated from Korean animals include the 394 residues of the N protein with subgroup A (Gyeonggi-do province) of Tyr and subgroup B (Gangwon-do province) of Cys [15]. The effects of substituting residue 394 in the KGH strain on immune response will require further investigation.
Two unique substitutions were observed in the P protein of the KGH strain. In rabies viruses, the N and P proteins are expressed, N–P complexes are formed, and viral genomic RNA is combined to inhibit N protein phosphorylation [44]. N phosphorylation is important for regulating viral RNA transcription and replication [45]. The P protein is present in two independent N-binding domains. One P protein functions as a chaperone by combining with newly formed non-RNA bound N protein (N0) at residues 69–177 of weaker N-binding sites, whereas another one is positioned at residues 268–297 and combines with N of the N-RNA complex [30, 46].
Ito et al. [32] confirmed the STAT1-binding domain at amino acid positions 267–297. Substitutions at residues 268 and 296 of the P protein in the KGH strain were found. Residue 268 is substituted from Ala to Gly only in the Korean and Indian isolates. Only residue 296 of the KGH strain was substituted from Thr to Val. Therefore, IFN production and its relationship with microtubule-dependent mechanisms may have been affected according to STAT1 regulation by substitution of these residues [47].
Takayama-Ito et al. [33] reported that the region at the position 164-303 of glycoprotein is related to pathogenicity in adult mice. Since then, another study has shown that glycoprotein 268 (Ile) residue plays a very important role in pathogenicity, and 242 (Ala), 255 (Asp), and 268 (Ile) may cooperate to the virulence of the Nishigahara strains [48]. Further studies are needed to elucidate the association of pathogenicity upon the substitution of glycoprotein 164 (Ile), 179 (Leu), and 245 (Ala) residues in KGH strain. No specific substitutions were observed in the M or L proteins.
According to Kuzmin et al. and Shao et al. [18, 19], the Arctic-like-1 clade is composed of regions in Iraq, Iran, Pakistan, Nepal, and India, whereas the Arctic-like-2 clade is composed of areas of south-eastern Siberia, Russian Far East, Japan, Inner Mongolia, and Korea. According to the complete genome sequence comparison in the present study, the Indian isolates of NNV-RAB-H and rabies virus serotype 1 (AY956319) were confirmed to have 91.6 % homology. In contrast, the G nt sequences of the rabies virus strain isolated in Korea have been confirmed to be more closely related to, and transmitted from, the NeiMeng1025C, NeiMeng1025B, NeiMeng927A, and NeiMeng927B isolates in northeastern China (Jilin province in China), than those in India (Fig. 2). Based on this result, China was verified as the primary source of Asian rabies events. Additionally, northern India and adjacent countries, such as Afghanistan, Nepal, Inner Mongolia, and Korea, have been confirmed to have common virus ancestors with the Arctic-like strain [16, 17, 49].
In Korea, animal rabies viruses that are found in limited areas of Gangwon-do and Gyeonggi-do provinces comprise four subgroups (Gangwon I, II, III, and Gyeonggi) [16]. Among the multiple animal rabies virus isolates, partial sequences of the N, P, and G genes as well as the G–L region have been reported [15, 50]. When the KGH strain was compared with the 23 glycoprotein gene sequence of rabies virus strains separated in Korea, the most closely related were determined to be SKRDG0204HC and SKRRD0205HC, which were isolated from dogs and raccoon dog in the Hwacheon area in 2002, around the same period when the patient was bitten (2001), and which belongs to the Gangwon III subgroup (Fig. 2). These results suggest that the rabies virus circulating in South and North Korea is a variety of viral variants, with features based on factors such as topography, animal species, and circulation year [18]. Although the complete genome sequence of Korean animal isolates is unknown, specific mutations in the N, P, and G genes were confirmed, and unique mutations were observed only in 342 nt (A to G) and 854 nt (C to T) in the G–L region.
In conclusion, we have provided the complete nucleotide and deduced amino acid sequence of a KGH rabies virus strain isolated from a Korean patient for the first time, and full sequence and genetic features of diverse rabies virus genomes were compared. In the N, P, and G protein functional region, the KGH strain was confirmed to have a unique substitution at sites related to the host immune response and pathogenicity. These results allow for an assessment of the interaction with host cells at the molecular level, and aid the evaluation of viral protein functions through direct manipulation of the viral genome. This result indicates that diverse genomic modifications occur among circulating rabies virus strains in other countries as well as in Korea. We expect that the complete genome analysis of the KGH strain will help us, not only to understand the phylogenetic relationships among rabies viruses strains all over the world, but also to select a candidate for vaccine development.
References
A.C. Jackson, W.H. Wunner, in Rabies, ed. by G.M. Baer, (Academic press, Elsevier, London, 2007)
H. Bourhy, J.-M. Reynes, E.J. Dunham, L. Dacheux, F. Larrous, V.T.Q. Huong, G. Xu, J. Yan, M.E.G. Miranda, E.C. Holmes, J. Gen. Virol. 89(Pt 11), 2673–2681 (2008)
E.C. Holmes, C.H. Woelk, R. Kassis, H. Bourhy, Virology 292, 247–257 (2002)
R.H. Fischman, F.E. Ward 3rd, Am. J. Epidemiol. 88(1), 132 (1968)
M.J. Schnell, J.P. McGettigan, C. Wirblich, A. Papaneri, Nat. Rev. Microbiol. 8, 51–61 (2010)
World Health Organization, WHO Expert consultation on rabies. World. Health. Organ. Tech. Rep. Ser. 931, 1–88 (2005)
A.R. Gould, A.D. Hyatt, R. Lunt, J.A. Kattenbelt, S. Hengstberger, S.D. Blacksell, Virus Res. 54, 165–187 (1998)
X. Wu, R. Franka, A. Velasco-Villa, C.E. Rupprecht, Virus Res. 129, 91–103 (2007)
T. Masatani, N. Ito, K. Shimizu, Y. Ito, K. Nakagawa, M. Abe, S. Yamaoka, M. Sugiyama, Virus Res. 155, 168–174 (2011)
L. Tao, J. Ge, X. Wang, H. Zhai, T. Hua, B. Zhao, D. Kong, C. Yang, H. Chen, Z. Bu, J. Virol. 84(17), 8926–8936 (2010)
Y. Kobayashi, Y. Suzuki, T. Itou, A.A.B. Carvalho, E.M.S. Cunha, F.H. Ito, T. Gojobori, T. Sakai, Infect. Genet. Evol. 10, 278–283 (2010)
Y.Z. Zhang, C.L. Xiong, X.D. Lin, D.J. Zhou, R.J. Jiang, Q.Y. Xiao, X.Y. Xie, X.X. Yu, Y.J. Tan, M.H. Li, Q.S. Ai, L.J. Zhang, Y. Zu, C. Huang, Z.F. Fu, Infect. Genet. Evol. 9, 87–96 (2009)
A.G. Szanto, S.A. Nadin-Davis, B.N. White, Virus Res. 136, 130–139 (2008)
T. Nagaraja, S. Madhusudana, A. Desai, Virus Genes 36, 449–459 (2008)
B.H. Hyun, K.K. Lee, I.J. Kim, K.W. Lee, H.J. Park, O.S. Lee, S.H. An, J.B. Lee, Virus Res. 114, 113–125 (2005)
D.K. Yang, Y.N. Park, G.S. Hong, H.K. Kang, Y.I. Oh, S.D. Cho, J.Y. Song, J. Vet. Sci. 12, 57–63 (2011)
S. Meng, Y. Sun, X. Wu, J. Tang, G. Xu, Y. Lei, J. Wu, J. Yan, X. Yang, C.E. Rupprecht, Virology 410, 403–409 (2011)
I.V. Kuzmin, G.J. Hughes, A.D. Botvinkin, S.G. Gribencha, C.E. Rupprecht, Epidemiol. Infect. 136, 509–519 (2008)
X.Q. Shao, X.J. Yan, G.L. Luo, H.L. Zhang, X.L. Chai, F.X. Wang, J.K. Wang, J.J. Zhao, W. Wu, S.P. Cheng, F.H. Yang, X.C. Qin, Y.Z. Zhang, Epidemiol. Infect. 139, 629–635 (2010)
J. Du, Q. Zhang, Q. Tang, H. Li, X. Tao, K. Morimoto, S.A. Nadin-Davis, G. Liang, Virus Res. 135, 260–266 (2008)
T. Masatani, N. Ito, K. Shimizu, Y. Ito, K. Nakagawa, Y. Sawaki, H. Koyama, M. Sugiyama, J. Virol. 84, 4002–4012 (2010)
B. Dietzschold, M. Lafon, H. Wang, L. Otvos Jr, E. Celis, W.H. Wunner, H. Koprowski, Virus Res. 8(2), 103–125 (1987)
H. Goto, N. Minamoto, H. Ito, N. Ito, M. Sugiyama, T. Kinjo, A. Kawai, J. Gen. Virol. 81, 119–127 (2000)
I.V. Kuzmin, L.A. Orciari, Y.T. Arai, J.S. Smith, C.A. Halon, Y. Kameoka, C.E. Rupprecht, Virus Res. 97, 65–79 (2003)
L. Geue, S. Schares, C. Schnick, J. Kliemt, A. Beckert, C. Freuling, F.J. Conraths, B. Hoffmann, R. Zanoni, D. Marston, L. McElhinney, N. Johnson, A.R. Fooks, N. Tordo, T. Müller, Vaccine 26, 3227–3235 (2008)
A.A.V. Albertini, A.K. Wernimont, T. Muziol, R.B.G. Ravelli, C.R. Clapier, G. Schoehn, W. Weissenhorn, R.W.H. Ruigrok, Science 313, 360–363 (2006)
M. Chenik, K. Chebli, Y. Gaudin, D. Blodel, J. Gen. Virol. 75, 2889–2896 (1994)
A.K. Gupta, D. Blondel, S. Choudhary, A.K. Banerjee, J. Virol. 74, 91–98 (2000)
O. Delmas, R. Assenberg, J.M. Grimes, H. Bourhy, RNA Biol. 7(3), 322–327 (2010)
G. Castel, M. Chtéoui, G. Caignard, C. Préhaud, S. Méhouas, E. Réal, C. Jallet, Y. Jacob, R.W.H. Ruigrok, N. Tordo, J. Virol. 83, 10808–10820 (2009)
D. Pasdeloup, N. Poisson, H. Raux, Y. Gaudin, R.W.H. Ruigrok, D. Blondel, Virology 334, 284–293 (2005)
N. Ito, G.W. Moseley, D. Blondel, K. Shimizu, C.L. Rowe, Y. Ito, T. Masatani, K. Nakagawa, D.A. Jans, M. Sugiyama, J. Virol. 84, 6699–6710 (2010)
M. Takayama-Ito, N. Ito, K. Yamada, N. Minamoto, M. Sugiyama, J. Neurovirol. 10, 131–135 (2004)
A. Guleria, M. Kiranmayi, R. Sreejith, K. Kumar, S.K. Sharma, S. Gupta, J. Biosci. 36, 929–937 (2011)
S. Finke, K.K. Conzelmann, J. Virol. 77, 12074–12082 (2003)
K. Shimizu, N. Ito, T. Mita, K. Yamada, J. Hosokawa-Muto, M. Sugiyama, N. Minamoto, Virus Res. 123, 154–160 (2007)
O. Poch, B.M. Blumberg, L. Bougueleret, N. Tordo, J. Gen. Virol. 71, 1153–1162 (1990)
D.A. Marston, L.M. McElhinney, N. Johnson, T. Müller, K–.K. Conzelmann, N. Tordo, A.R. Fooks, J. Gen. Virol. 88, 1302–1314 (2007)
I.V. Kuzmin, X. Wu, N. Tordo, C.E. Rupprecht, Virus Res. 136, 81–90 (2008)
M.A. McClure, J. Perrault, Virology 172, 391–397 (1989)
P. Ming, J. Du, Q. Tang, J. Yan, S.A. Nadin-Davis, H. Li, X. Tao, Y. Huang, R. Hu, G. Liang, Virus Res. 143, 6–14 (2009)
M.J. Schnell, T. Mebatsion, K.K. Conzelmann, EMBO J. 13, 4195–4203 (1994)
K. Honda, T. Taniguchi, Nat. Rev. Immunol. 6, 644–658 (2006)
P. Liu, J. Yang, X. Wu, Z.F. Fu, J. Gen. Virol. 85, 3725–3734 (2004)
X. Wu, X. Gong, H.D. Foley, M.J. Schnell, Z.F. Fu, J. Virol. 76, 4153–4161 (2002)
S. Finke, K. Brzóka, K.K. Conzelmann, J. Virol. 78, 12333–12343 (2004)
G.W. Moseley, X. Lahaye, D.M. Roth, S. Oksayan, R.P. Filmer, C.L. Rowe, D. Blondel, D.A. Jans, J. Cell Sci. 122, 3652–3662 (2009)
M. Takayama-Ito, N. Ito, K. Yamada, M. Sugiyama, N. Minamoto, Virus Res. 115, 169–175 (2006)
T. Matsumoto, K. Ahmed, O. Wimalaratne, K. Yamada, S. Nanayakkara, D. Perera, D. Karunanayake, A. Nishizono, Arch. Virol. 156, 659–669 (2011)
Y.J. Park, M.K. Shin, H.M. Kwon, Virus Genes 30, 341–347 (2005)
Acknowledgments
This study supported by grant no. 2010-N53001-00 from the Korea National Institute of Health.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Park, JS., Kim, CK., Kim, S.Y. et al. Molecular characterization of KGH, the first human isolate of rabies virus in Korea. Virus Genes 46, 231–241 (2013). https://doi.org/10.1007/s11262-012-0850-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0850-6