
Abstract
Equine rhinitis A virus (ERAV) is an ubiquitous virus, routinely identified in equine respiratory infections; however, its role in disease and genetic features are not well defined due to a lack of genomic characterization of the recovered isolates. Therefore, we sequenced the full-length genome of a Canadian ERAV (ERAV/ON/05) and compared it with other ERAV sequences currently available in GenBank. The ERAV/ON/05 genome is 7,839 nucleotides (nts) in length with a variable 5′UTR and a more conserved 3′UTR. When ERAV/ON/05 was compared to other reported ERAV isolates, an insertion of 13 nt in the 5′UTR was identified. Further phylogenetic analysis demonstrated that ERAV/ON/05 is closely related to the ERAV/PERV isolate, which was isolated in 1962 in the United Kingdom. The polyprotein of ERAV/ON/05 had a 96 % nucleotide and amino acid sequence identity to reported ERAVs, and it appears that, despite the high error rate of RNA-dependent RNA polymerase, this isolate has retained high sequence identity to the strain first described by Plummer in 1962.
Similar content being viewed by others

Introduction
It is estimated that horses are exposed to equine rhinitis A virus (ERAV) by the time they reach their second year of age [1, 2]. As a respiratory virus, ERAV has been associated with upper respiratory disease in horses [3, 4]; however, its clinical significance as a primary agent in respiratory disease remains controversial. Recently we have found ERAV to be highly prevalent among horses in Ontario during respiratory outbreaks [5]. Generally, virus recovery from most clinical cases is unsuccessful, leaving retrospective serology and reverse transcriptase-PCR (RT-PCR) as the only tools for confirmation of infection. Moreover, ERAV non-cytopathic strains have been identified in the equine population [3], making its diagnosis challenging. Thus, due to a low viral recovery rate, only a few complete genome sequences are currently available. Human rhinovirus (HRV) is among the leading causes of respiratory infections in humans [6], and more than 100 serotypes are recognized at this time [7]. In contrast, in the equine species only two genera (ERAV and ERBV) with four serotypes (ERAV, ERBV1, ERBV2, and ERBV3) are currently known [8, 9].
Equine rhinitis A virus, previously known as Equine rhinovirus 1 [9], was first described in the horse in 1962 [10]. Sequence analysis of ERAV has demonstrated that its genetic characteristics make it closely related to foot and mouth disease virus (FMDV) [11, 12]. ERAV is one of four species in the genus Aphthovirus, family Picornaviridae, along with FMDV, bovine rhinitis A virus, and bovine rhinitis B virus [9]. While ERAV has been allocated to the genus Aphthovirus, the equine rhinitis B virus serotypes ERBV1, ERBV2, and ERBV3 are included in the genus Erbovirus within the family. Viruses in this family are not enveloped, with a capsid composed of four structural viral proteins (VP1, VP2, VP3, and VP4) forming a protomer. The picornavirus genome is a positive single strand of RNA with a size that varies from 7.0 to 8.0 kb encoding a single polyprotein that includes the structural and non-structural proteins [13]. The polyprotein coding region is flanked by a long (up to 1,200 nt) 5′ untranslated region (5′UTR) and a short 3′ untranslated region (3′UTR) [9]. Unfortunately, most of these characteristics have been identified only in other members of the family Picornaviridae due to a low ERAV recovery rate.
Recently, we recovered an ERAV isolate (ERAV/ON/05) from a nasopharyngeal swab collected from an equine respiratory outbreak in Ontario, Canada [5]. The acute respiratory disease in these horses was characterized by pyrexia, nasal discharge, and cough; signs, which are not virus-specific. ERAV was first identified in North America in 1962; however, viral recovery and genomic characterization from North America has not been routinely attempted. Therefore, the objective of this study was to characterize the genome of the ERAV/ON/05 isolate.
Materials and methods
Cells and virus
Rabbit kidney-13 (RK-13) cells were grown in Dulbecco’s modified Eagle’s medium nutrient mixture F12 HAM (Sigma-Aldrich Canada Ltd., Oakville, Ontario) with 2–5 % fetal bovine serum (Sigma-Aldrich Canada Ltd., Oakville, Ontario). The ERAV/ON/05 isolate (second passage) (GenBank accession number: JX294351) was propagated in RK-13 cells and aliquots were stored at −70 °C for later work. RK-13 monolayers were inoculated at 90 % confluence and RNA was extracted approximately 8–12 h post-infection (p.i.), before cytopathic effect was observed.
Virus titration and growth kinetics
A plaque assay was used to titrate all samples during these experiments. Briefly, RK-13 cells were grown in 3 cm diameter Petri dishes and infected with ERAV/ON/05 at 90 % confluence. Adsorption was allowed for 45 min and the inoculum was removed and replaced with a 0.7 % agarose layer. Plates were checked every 12 h and the plaques were counted and classified as small and large. The viral titer was calculated from the number of plaques at the highest dilution and recorded as a plaque-forming unit. For plaque purification, five plaques of each size (small and large) were picked into 300 μl of medium and frozen at −70 °C. Viruses from each plaque were propagated in RK-13 cells, and RNA was extracted for sequence comparison of the 5′UTR. The latter was to determine if plaque size was associated with genomic changes in this region. In order to study the growth characteristics of ERAV/ON/05, RK-13 cells were infected with a multiplicity of infection of 5. The cells were incubated at 37 °C and samples were collected every 4 h for a period of 28 h. All samples were titrated by the plaque assay and the results were plotted to construct a one-step growth curve.
RNA extraction and sequencing
RNA was extracted from ERAV/ON/05-infected RK-13 cells. Cells were treated with 1 ml of TRIzol (Invitrogen Canada Inc., Burlington, ON) at 8–12 h p.i. and extraction was performed according to the manufacturer’s recommendations. RNA pellets were eluted in 30 μl of RNAse-free water and kept at −70 °C. The first strand cDNA was synthesized using SuperScript™ II Reverse Transcriptase and random primers (Invitrogen Canada Inc., Burlington, ON) following the manufacturer’s recommendations. A 50 μl PCR reaction was carried out using Taq DNA polymerase (Invitrogen Canada Inc., Burlington, ON) and a set of sense and antisense primers (Table 1). The genome of this isolate was amplified by the primer walking approach, and primer design was based on seven ERAV sequences available in GenBank. The PCR conditions were: 4 min at 94 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 55 °C, and 30 s at 72 °C with a final extension at 72 °C for 10 min. Sequencing of the 5′ and 3′ ends were completed with the 5′ RACE and 3′ RACE kits (Invitrogen Canada Inc., Burlington, ON) as recommended by the manufacturer. Several nested PCRs were required to amplify the 5′UTR end.
Genome analysis
Preliminary identification of the virus was done by sequencing of the viral protein 1 gene (VP1) using primers derived from sequences available in GenBank. Sequencing reactions were set and run by the Laboratory Services Division at the University of Guelph. All primers were designed on Gene Runner version 3.05 (Hastings Software Inc., Hastings, NY). The sequences were assembled and edited using EditSeq and SeqMan DNASTAR Lasergene 8 (DNASTAR Inc., Madison, WI). Sequencing results were entered into the BLAST software [National Center for Biotechnology Information, Bethesda, MD (NCBI)] and compared to similar entries in GenBank. ClustalW2 [European Bioinformatics Institute, Dublin, Ireland (EBI)] [14] was used for multiple sequence alignment and preliminary construction of the phylogenetic tree. The final phylogenetic tree was created on MEGA 5.0 by the Maximum Composite Likelihood method and the reliability was evaluated by bootstrapping with 1,000 replications [15]. Analysis of the nucleotide sequences was plotted on SimPlot Version 3.5.1 (Baltimore, MD, USA). In order to investigate the possibility of viral recombination between ERAV isolates, we completed a Bootscan analysis on SimPlot (Version 3.5.1) comparing the genomic sequences of all complete ERAV available in GenBank (7 reports) and ERAV/ON/05. Polyprotein cleavage sites were predicted based on the genome sequence of the original ERAV from 1962 available in GenBank (accession numbers: DQ272578 and NC003982). In addition, the RNA secondary structures from the 5′UTR were predicted using the mFOLD software [16].
Results
Characterization of ERAV/ON/05
ERAV/ON/05 was recovered from a febrile horse in Ontario, Canada [5]. This isolate was propagated in RK-13 cells and titrated by plaque assay. As for other picornaviruses, in vitro growth of the ERAV/ON/05 isolate showed an increase in titer at 4 h p.i., reaching a plateau by 12 h p.i., during the one-step growth curve study. RK-13 cell monolayers were almost completely destroyed at 28 h p.i. Microscopically, infection was detected by cell rounding and detachment from the monolayer. Plaques seen in these cells during ERAV/ON/05 infection varied in shape and size with diameters ranging from 2 to 7 mm (Fig. 1).

Plaque morphology of ERAV/ON/05 in RK-13 cells. Top left box is a photograph of control RK-13 cells. Remaining boxes are photographs of ERAV/ON/05 infected RK-13 cells, showing a variety of small and large plaques within the same plate
Genome sequence analysis
Nucleic acids coding for the VP1, were amplified, sequenced, and compared to seven sequences in GenBank [accession numbers: NC003982 (UK), DQ272577 (USA), DQ272128 (USA), DQ272127 (USA), DQ268580 (USA), DQ272578 (UK), and L43052 (UK)]. Results from this initial comparison demonstrated a maximum amino acid identity of 95 % to ERAV VP1 from viruses recovered in the UK and USA. The complete genome of the ERAV/ON/05 isolate was 7,839 nt in length with a GC content of 47 % including the poly (A) tail. Four identical 20-nt-repeats (CTGTAGCGTCAGTAAAACGC) separated by 18, 21, and 18 nt were identified at the 5′UTR. The ERAV/ON/05 5′UTR was composed of 940 nt with a 54 % GC content. Interestingly, various insertions (three-1, two-2, and one-13 nt) and four small deletions (two-1, one-2, and one-3 nt) in the 5′UTR were identified (Fig. 2). mFOLD analysis of 5′UTR of the ERAV/ON/05 showed that these insertions and deletions were located near the poly (C) tract on the first predicted stem loops of the internal ribosome entry site (IRES). Alignment of the 5′UTR sequence of all available ERAV demonstrated a lower identity, ranging from 73 to 81 %, making the 5′UTR of the ERAV/ON/05 isolate the most variable region. No other major changes were observed throughout the entire genome. RK-13 cell infection with ERAV/ON/05 yielded a mixture of plaque sizes. Sequence analysis of a 426 nt fragment at the 5′UTR of five small and five large plaques showed no nucleotide differences in this region among plaques.

ClustalW alignment of the Ontario ERAV (ERAV/ON/05) and Plummer’s original ERAV isolate from 1962 (PERV-1). The alignment shows nucleotide insertions (black) and deletions (gray) in the 5′UTR of ERAV/ON/05
A single polyprotein ORF with 6,747 nt (2,248 amino acids) with 46.8 % GC content was identified. The translation initiation site was detected at nt 940 with the AUG start codon and ending at nt 7686 with the UAA stop codon. A subsequent AUGAUG sequence was identified 58-nt downstream from the polyprotein start codon. Analysis with Blastx and Blastp (NCBI) of the nucleotide and amino acid sequences of the polyprotein of this isolate showed a 96 % identity with other reported ERAVs. Amino acid sequence analysis revealed an identical protein (structural and non-structural) arrangement and length along the entire genome. These comparisons were made with PERV and PERV-1 reported genome sequences (accession numbers NC003982 and DQ272578, respectively), which correspond to the genomic characterization made by two different research groups using Plummer’s isolate from 1962 [12, 17].
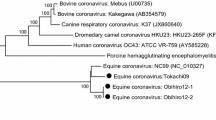
The 3′UTR was composed of 110 nt with a 24.3 % GC content, and a poly-A tail. Alignments of the 3′UTR sequences of all available ERAV, demonstrated that the identity ranged from 75 to 81 %. SimPlot analysis showed high nucleotide similarity (percentage) among all ERAV isolates and ERAV/ON/05 (Fig. 3a). Major disparities were identified between nt 1250 and 1750 and between nt 4500 and 4750. Nevertheless, this analysis showed that nucleotide similarity among all reported isolates was between 70 and 82 %. In order to further investigate the divergence in the genomes, a scan (Bootscan) to identify possible recombination was performed (Fig. 3b). These analyses demonstrated that the Ontario isolate did not have predicted recombination sites with other ERAV isolates. Nevertheless, when the Ontario isolate was removed from the analysis, there was evidence suggesting possible recombination between other ERAV isolates in the past (data not shown). As expected, the full-length nucleotide sequence of ERAV/ON/05 falls within other picornaviruses and is closely related to FMDV (Fig. 4a). Phylogenetic analysis showed that ERAV/ON/05 clusters with Plummer’s UK isolate from 1962 (Plummer [10]) despite the few nucleotide changes found (GenBank accession number: DQ272578) (Fig. 4b).

Equine rhinitis A virus sequences available in GenBank are presented as a percentage (SimPlot Version 3.5.1). a Similarity scores of complete genome sequences of ERAV are shown. The Ontario ERAV isolate (ERAV/ON/05) was used as a query and compared to ERAVs in GenBank under accession numbers: NC003982, DQ272578, DQ272127, L43052, DQ272128, DQ268580, and DQ272577. The horizontal bar above the plot is a cartoon of the ERAV genome. b Bootscan analysis of ERAVs available in GenBank including ERAV/ON/05

Phylogenetic analysis of the Ontario ERAV (ERAV/ON/05) full-length genome. Bootstrap neighbor-joining tree was designed using MEGA 5. Reliability was evaluated by bootstrapping with 1,000 replications. a Phylogenetic comparison of ERAV/ON/05 to other ERAV, ERBVs, FMDV, and HRV. b Phylogenetic comparison of all ERAV sequences currently available in GenBank
Discussion
This is the first Canadian ERAV genome that has been sequenced and analyzed. ERAV is not routinely sought and recovered from clinical cases during equine respiratory outbreaks; therefore, isolation of ERAV has been incidental in most cases. Previously, equine rhinitis viruses were classified within the family Picornaviridae, but were not clearly assigned to a specific genus. In 1996 Li et al. [11] and Wutz et al. [12] demonstrated that, on the basis of phylogenetic characteristics, ERAV was closely related to FMDV. In this study, we found ERAV/ON/05 to be closely related to the reported North American and European ERAV isolates, including the first ERAV ever recovered. This close relationship between ERAV/ON/05 and the 1962 isolate indicates that 50 years after the first ERAV was reported, the viral genome of ERAV/ON/05, and more specifically the polyprotein amino acid sequence, has remained largely unchanged. This suggests that the few small modifications detected at the nucleotide level do not compromise in vitro or in vivo viral replication and might only reflect the normal genetic evolution of this virus. However, it is not clear if these few nucleotide changes affect virulence, and further studies should be conducted.
Unexpectedly, it appears that ERAV has been able to maintain its genomic characteristics for over 50 years, as demonstrated by identity between the genome sequences of ERAV/ON/05 and Plummer’s 1962 isolate. Environmental changes and possibly host immunomodulation have played an important role in disease control; however, these factors have not been sufficient to induce major genomic changes. In contrast, HRV has been shown to undergo genomic mutations over time, which in combination with a high number of circulating serotypes makes vaccine development and human disease control an unattainable task [7].
Our isolate was 7,839 nt in length, including the 5′UTR, the polyprotein gene, 3′UTR, and the poly-A tail. The genome sequence of ERAV/ON/05 is one of the few complete ERAV sequences reported to date. It is important to note that our data showed the lowest identity scores in the 5′UTR and 3′UTR regions and this may have been due to a lack of sequencing data from reported isolates. The 5′UTR sequence revealed the presence of three repeats that have been previously described in other ERAV and commonly found on the FMDV [12, 18]. It has been suggested that these repeats may be required in the formation of the secondary structures found on the 5′UTR, the IRES [19]. The identity analysis showed that the polyprotein of ERAV/ON/05 bears highly conserved nucleotide sequences. As an RNA virus, ERAV is prone to constant mutations due to a lack of proofreading by the polymerase. Even though this RNA virus has been under natural evolution and constant replication, no significant genomic changes have been introduced since its first recovery. It is evident that the structural and non-structural proteins, which are encoded within the polyprotein, represent the most conserved regions in the genome. As reported elsewhere [20], the VP1 protein was shown to be highly conserved, and could be considered as a diagnostic target region as recommended by others [20–22]. Additionally, there is the opportunity for an effective vaccine to be developed since the ERAV isolates recovered and identified are similar, irrespective of time and location.
Viral replication in cell culture was demonstrated to be rapid and efficient. A complete viral replication cycle was detected by 4 h, reaching a plateau in 12 h. A short and proficient replication cycle is a common characteristic of picornaviruses and more specifically of HRV [23], which may reflect the viral activity in vivo. Such features might explain the severity and speed of clinical respiratory signs in the natural host (equines). For other picornaviruses, such as poliovirus, the presence of small deletions and/or insertions in the 5′UTR have been associated with differences in plaque size in cell culture, and increased virulence in vivo [24]. Although, we found that ERAV/ON/05 generated various plaque sizes in RK-13 cells, analysis of the 5′UTR nt sequences of viruses from several plaques found no differences between small and large plaques. This suggests that variation in plaque size in cell culture may be due to a cell growth characteristic (e.g., stage of the cell cycle), rather than an association between viral replication and the nucleotide sequence in the 5′UTR of ERAV/ON/05.
In summary, our findings confirm that ERAV/ON/05 is closely related to the ERAVs sequenced and reported to date. Interestingly, it appears that the ERAV genome has not changed significantly over the past 50 years and only small nt changes were identified in the 5′UTR of the ERAV/ON/05 isolate. These changes may be incidental or reflect minor adaption to its natural host, or a small degree of adaptation to other environmental changes. Further studies to determine the evolutionary pattern of ERAV and establish a correlation between geographical isolation and genomic characteristics are required.
References
J. Ditchfield, L.W. Macpherson, Cornell Vet. 55, 181 (1965)
R. Willoughby, L. Huber, L. Viel, in Proceedings from the 7th ACVIM forum, San Diego, 1989, p. 604
F. Li, H.E. Drummer, N. Ficorilli, M.J. Studdert, B.S. Crabb, J. Clin. Microbiol. 35, 4 (1997)
M. Klaey, M. Sanchez-Higgins, D.P. Leadon, A. Cullinane, R. Straub, H. Gerber, Equine Vet. J. 30, 3 (1998)
A. Diaz-Mendez, L. Viel, J. Hewson, P. Doig, S. Carman, T. Chambers, A. Tiwari, C. Dewey, Can. J. Vet. Res. 74, 4 (2010)
P. McErlean, L.A. Shackelton, S.B. Lambert, M.D. Nissen, T.P. Sloots, I.M. Mackay, J. Clin. Virol. 39, 2 (2007)
A.C. Palmenberg, D. Spiro, R. Kuzmickas, S. Wang, A. Djikeng, J.A. Rathe, C.M. Fraser-Liggett, S.B. Liggett, Science 324, 5923 (2009)
W.D. Black, C.A. Hartley, N.P. Ficorilli, M.J. Studdert, J. Gen. Virol. 86(Pt 8), 2323 (2005)
N.J. Knowles, T. Hovi, T. Hyypiä, A.M.Q. King, M. Lindberg, M.A. Pallansch, A.C. Palmenberg, P. Simmonds, T. Skern, G. Stanway, T. Yamashita, R. Zell, in Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses, ed. by A.M.Q. King, M.J. Adams, E.B. Carstens, E.J. Lefkowitz (Elsevier, San Diego, 2011), pp. 855–880
G. Plummer, Nature 195(4840), 519 (1962)
F. Li, G.F. Browning, M.J. Studdert, B.S. Crabb, Proc. Natl. Acad. Sci. USA. 93, 3 (1996)
G. Wutz, H. Auer, N. Nowotny, B. Grosse, T. Skern, E. Kuechler, J. Gen. Virol. 77, 8 (1996)
V. Racaniello, Picornaviridae: The Viruses and their Replication (Lippincott Williams & Wilkins, Philadelphia, 2007), p. 795
M.A. Larkin, G. Blackshields, N.P. Brown, R. Chenna, P.A. McGettigan, H. McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J. Gibson, D.G. Higgins, Bioinformatics 23, 21 (2007)
K. Tamura, J. Dudley, M. Nei, S. Kumar, Mol. Biol. Evol. 24, 8 (2007)
M. Zuker, Nucleic Acids Res. 31, 13 (2003)
A. Aminev, A. Palmenberg, Comparative Genomics Among Equine Rhinitis A Viruses (Unpublished, University of Wisconsin-Madison, Madison, WI, 2005)
S. Forss, H. Schaller, Nucleic Acids Res. 10, 20 (1982)
T.M. Hinton, F. Li, B.S. Crabb, J. Virol. 74, 24 (2000)
R.A. Stevenson, C.A. Hartley, J.A. Huang, M.J. Studdert, B.S. Crabb, S. Warner, J. Gen. Virol. 84(Pt 6), 1607 (2003)
C.A. Hartley, N. Ficorilli, K. Dynon, H.E. Drummer, J.A. Huang, M.J. Studdert, J. Gen. Virol. 82(Pt 7), 1725 (2001)
S. Warner, C.A. Hartley, R.A. Stevenson, N. Ficorilli, A. Varrasso, M.J. Studdert, B.S. Crabb, J. Virol. 75, 19 (2001)
S. Todd, J.S. Towner, B.L. Semler, Virology 229, 1 (1997)
N. Hamada, Y. Imamura, M. Shingu, J. Med. Virol. 24, 1 (1988)
Acknowledgments
This study would not have been possible without the generous support of Boehringer Ingelheim (Canada) Ltd., Vetmedica, Burlington, Ontario, the EP Taylor Equine Research Fund and the Equine Guelph Research Program.
Author information
Authors and Affiliations
Corresponding author
Additional information
GenBank: JX294351.
Rights and permissions
About this article
Cite this article
Diaz-Méndez, A., Viel, L., Shewen, P. et al. Genomic analysis of a Canadian equine rhinitis A virus reveals low diversity among field isolates. Virus Genes 46, 280–286 (2013). https://doi.org/10.1007/s11262-012-0848-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0848-0