
Abstract
Six clinical cases of avipoxvirus (APV) infection were investigated and molecular biologically studied. The samples were collected from different domesticated birds reared in the Egyptian backyard management system and were propagated on the chorioallantoic membrane of embryonated chicken eggs. The virus isolation was confirmed via PCR amplification of fpv167 (P4b) gene locus. All the studied isolates were characterized as Fowlpox-like viruses based on the amplicon length of fpv140 gene locus. The phylogenetic analysis of fpv167 (P4b) gene clustered Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV strains within subclade A1. Furthermore, Elsharqyia_PGPV strain was clustered within subclade A2 (Turkeypox virus) and showed 100 % nucleic acid identity with the wood pigeon Indian which was isolated in 2009. On the other hand, when the fpv140 gene was used for the phylogenetic analysis, Elsharqyia_PGPV was clustered within subclade A4 (Pigeonpox virus) with the other PGPVs. This study is considered the first molecular record for APVs circulating in the Egyptian birds. Further studies in a larger scale need to be developed to have a better understanding about the molecular characterization of the Egyptian APV strains.
Similar content being viewed by others


Introduction
The Poxviridae family is subdivided into the entomopoxvirinae and chordopoxvirinae subfamilies. The genus avipoxvirus (APV) has the largest and the most divergent genome among the chordopoxvirus genera [8]. It has many species such as Fowlpox virus (FWPV), Turkeypox virus (TKPV), Pigeonpox virus (PGPV), Canarypox virus (CNPV), Ostrichpox virus (OSPV), Penguinpox virus (PEPV), Falconpox virus (FLPV), and Sparrowpox virus (SRPV), and many others recently emerged poxviruses in different avian species. There are about 9,000 bird species; 232 species of them have been recorded to have acquired a natural poxvirus infection and to have various forms of poxvirus infection [4]. Avian pox is a slowly spreading disease characterized by the development of discrete proliferative nodular skin lesions (cutaneous form) or fibrino-necrotic lesion in the mucous membrane of the upper respiratory tract (diphtheritic form) [19]. APV infection is usually associated with a low mortality rate in chickens and turkeys; however, during some APV outbreaks, the mortality rate can reach 65–100 %, especially in the Canary [18, 19]. FWPV has a double stranded DNA genome which contains a central coding region surrounded by two identical inverted terminal repeat regions. The genome size is 288 kbp approximately and encodes 260 open reading frames, of which 101 exhibit similarity to genes of known function [1]. fpv167 is a conserved gene among APVs that encodes orthologs of vaccinia virus core protein P4b. Polymerase chain reaction amplification of the fpv167 specific 578-bp fragment was applied to diagnose APV infection [10]. Lüschow et al. [12] found that fpv167 (P4b) PCR amplification in combination with restriction endonuclease analysis and sequencing are rapid and effective diagnostic tools for APVs. Although it has been a successful diagnostic marker, PCR amplification of the P4b gene could not distinguish between different APV strains. However, sequencing and phylogenetic analysis of fpv167 (P4b) nucleotide sequence discriminate between different APV strains and revealed divergence among the different viruses that can be consistently correlated to the host species [21]. fpv140, an ortholog of vaccinia virus strain Copenhagen H3L, was also used in PCR amplification of APVs and interestingly it was useful in the differentiation between Fowlpox-like and Canarypox-like viruses on the basis of their fragment size. However, the routine usage of this gene for APV molecular characterization has some limitations. Since PCR of fpv140 was carried out for 15 isolates in northern Italy, eventually only seven of them gave positive results [13, 16].
Based on the phylogenetic analysis of fpv167 (P4b), APVs are divided into three clades; clade A (FWPV), clade B (CNPV), and clade C (Psittacinepox virus) [11, 13]. Jarmin et al. suggested additional nomenclature for the phylogenetic groups; FWPV (subclade A1), TKPV (subclade A2), Falcon and Albastrosspox virus (subclade A3), CNPV (subclade B1), SLPV (subclade B2), and PSPV (clade C). Although according to the phylogenetic analysis of fpv167 (P4b) PGPV could not be used as a specific nomenclature as it can be clustered in either subclade A2 or B2 [11], it has a specific nomenclature and can be distinguished from the TKPV and SLPV in a separate cluster (subclade A4) based on the phylogenetic analysis of fpv140 (H3L) nucleotide sequence [11].
Many studies have been designed to analyze different APVs from different geographic areas and study their phylogenetic relationship with each other. One such example, in northern Italy, sequencing analysis of 15 isolates revealed that most of the isolates belonged to either clade A or clade B, whereas only one isolate from Japanese quail was classified and clustered with clade C [11, 13]. Another example, the majority of New Zealand APV isolates belonged to A1 subclade; however, an isolate from a wood pigeon (kereru) belonged to subclade A3 [9]. In India, APV infection has been reported in different wild and domesticated birds [3, 14, 15]. The authors explained the recurring of FWPV infection in the backyard Indian chickens due to the integration of a near full-length reticuloendotheliosis provirus in the FWPV genome which leads to state of immunosuppression [3].
In Egypt, there are many reports discussing the isolation and the epidemiological studies of APVs [2, 6, 7, 17]. So far and up till now, there are no available data about the molecular characterization of APVs in Egypt and their phylogenetic relationship with each other and the other published strains in the database. In the present study, we provide the first report for the sequence analysis of the fpv167 (P4b) and fpv140 loci amplified from different APVs. Six isolates were recovered from different domesticated backyard bird species in the Elsharqyia province, 2011.
Results
Virus isolation
Virus isolation was performed on embryonated chicken eggs (ECE) obtained from non-vaccinated chicken flock. Pock lesions were evident on CAM after either the first (Elsharqyia_TKPV, Elsharqyia_PGPV, Elsharqyia_FWPV2, Elsharqyia_FWPV3, and Elsharqyia_FWPV4 isolates) or second (Elsharqyia_FWPV1) passage. Except Elsharqyia_PGPV, morphologically, the pocks were similar for most of the samples with grayish-white discoloration, compact shape, and marked thickening of the CAM. Elsharqyia_PGPV was isolated from the pigeon clinical sample and the pocks have a characteristic morphological shape with yellowish discoloration, nodular shape, and moderate thickening of the CAM.
Histopathology
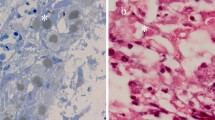
Histopathologic examination of the cutaneous lesions revealed ballooning degeneration of the keratinocytes having large eosinophilic intracytoplasmic inclusions with central pale zone (Bollinger bodies). The virus replication evidences were observed in the CAM consisted of hyperplasia of the epithelium with cellular edema (hydropic degeneration) and Bollinger bodies (Fig. 1).

Histopathology of APV lesions. Histopathology of CAM of ECE inoculated with APV isolate (Elsharqyia_PGPV) which was collected from Elsharqyia province in 2011 showing cellular edema (hydropic degeneration) with eosinophilic intracytoplasmic Bollinger bodies (indicated by arrows)
PCR and sequencing
PCR was performed for vaccinal strain (FWPFPV9) and skin lesions as well as pock lesions from CAMs. Two PCRs were performed with two sets of primers for the detection of APV-specific DNA (P4b-578 bp) and then discrimination between Fowlpox-like viruses and Canarypox-like viruses (fpv140 gene). In the first PCR, avian Poxvirus-specific 578-bp fragment was detected from the vaccinal strain (FWPVFPV9) and all the tested skin & CAM samples (Fig. 2a). All the amplicons from the second PCR were clustered as FWPV-specific with 1,800 bp (Fig. 2b). Sequencing of fpv167-578-bp and fpv140-1,800-bp fragments were carried out for six isolates which were named as Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, Elsharqyia_TKPV, and Elsharqyia_PGPV. The obtained nucleotide sequences were submitted to the GenBank under accession number (Table 1).

a PCR amplification of the fpv167 (P4b) gene from APV isolates of the present study. Lane M 1-kbp DNA ladder (Fermentas), CTRL− negative control and CTRL+ positive control (FWPVFP9). See Tables 1 and 2 for virus abbreviations. b PCR amplification of the fpv140 (H3L) gene from APV isolates of the present study. Lane M 1 kbp DNA ladder (Fermentas), CTRL− negative control and CTRL+ positive control (FWPVFP9). See Tables 1 and 2 for virus abbreviations
Alignment and phylogenetic analysis of fpv167 (P4b) gene
The amino acid sequence alignment analysis of one hundred sixty-eight amino acids (truncated to have the same length) of the six isolates was performed using CLC main workbench. The obtained sequences were aligned with the published APVs-P4b amino acid sequence of strains FWPVFPV9, TKPV13401, and PGPVP with the accession numbers AJ581527, AY530304, and AM050385, respectively, which were used as reference strains. The alignment analysis showed that 100 % amino acid identity between the Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV strains compared to FWPVFPV9 and TKPV13401. Consistently, Elsharqyia_PGPV showed 100 % amino acid identity compared to PGPVP as a reference strain. On the other hand, the comparative alignment of Elsharqyia_PGPV revealed 98.2 % amino acid sequence identity to Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV (S1).
For the phylogenetic analysis of the studied strains, the nucleotide sequences of 37 strains isolated from 16 different bird species were aligned (Table 2). The phylogenetic analysis was performed for 428 truncated sequences by neighbor-joining (N-J) analysis with bootstrapping (1000). As previously shown, the DNA tree of APVs branched into three major clades: A (Fowlpox-like viruses), B (Canarypox-like viruses), and C (Psittacinepox-like viruses). Clade A is further subdivided into subclade A1, A2, A3, and A4, whereas clade B is subdivided into subclade B1 and B2. The five strains, Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV, were clustered in subclade A1 and showed 100 % nucleotide identity with the other FWPV strains in addition to TKPV13401 and SRPVDD1258 strains. Viruses in subclade A1 showed 90.89, 91.82, and 88.32 % nucleotide identity to the other viruses in subclade A2, A3, and A4, respectively. Furthermore, they showed an average of 76.6 and 75.85 % nucleotide identity to CNPV-like group of viruses (Clade B) and Psittacine viruses (Clade C), respectively. Elsharqyia_PGPV strain was grouped in subclade A2 together with the other TKPVs (TKPV66 and TKPV98), in addition to PGPVP, PGPVTP-2, PEPV, and OSPV strains. It showed 90.89, 100, 97.66, and 89 % nucleotide identity to the viruses in subclade A1, A2, A3, and A4, respectively. In addition, it showed an average of 75.7 % nucleotide identity to the Canarypox-like virus isolates (Clade B) and 75.15 % nucleotide identity to Psittacinepox-like virus isolates (Clade C) (Fig. 3).

Phylogenetic analysis of APVs based on fpv167 (P4b) gene nucleotide sequences. Phylogenetic tree was constructed via multiple alignments of 428-bp nucleotide sequence of P4b gene from 43 APV strains. The tree was analyzed by neighbor-joining (N-J) analysis with bootstrapping (1000). APV clades and subclades are labeled. (Asterisk) Elsharqyia_FWPV label represents Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, and Elsharqyia_FWPV4. See Tables 1 and 2 for virus abbreviations
Alignment and phylogenetic analysis of fpv140 (H3L) gene
The alignment analysis of the full length amino acid sequences of fpv140 (H3L) gene from the six isolates was performed using CLC main workbench. The obtained sequences were aligned together and with the published APV-H3L amino acid sequence of strains FWPVFPV9, TKPV66, and PGPVP with the accession numbers AJ581527, AM071390, and AM071389, respectively. The alignment analysis revealed 100 % amino acid identity between the Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV isolates. The five isolates showed 99.7 and 87.13 % amino acid identity when they were aligned with the reference strains FWPVFPV9 and TKPV66, respectively. Elsharqyia_PGPV showed 84.90 % amino acid identity compared to PGPVP as a reference strain. On the other hand, the comparative alignment of Elsharqyia_PGPV revealed 74.26 % amino acid identity to Elsharqyia_FWPVs and Elsharqyia_TKPV. Remarkably, insertion of 13 amino acid residues (FILSSYVL amino acids 279–286) and (WEIRY amino acids 293–297) were observed in Elsharqyia_PGPV-fpv140 and were not present in PGPVP strain (S2). In addition, Elsharqyia_PGPV-fpv140 has a deletion in 9 amino acid residues (GLAIFDVNN amino acids 310–318) which are present in PGPVP strain.
For the phylogenetic analysis of the studied strains, the nucleotide sequences of 16 strains isolated from eight different bird species were aligned (Table 2). The phylogenetic analysis based on fpv140 gene sequences was performed for 896 truncated sequences by neighbor-joining (N-J) analysis with bootstrapping (1000). As previously shown [11], the DNA tree of APVs based on fpv140 gene sequences branched into two major clades: A and B. Clade A is further subdivided into subclade A1 (Fowlpox virus), A2 (TKPV), A3 (Falcon and Albastrosspox Virus), and A4 (PGPV), whereas clade B is a Canarypox virus. Elsharqyia_FWPVs and Elsharqyia_TKPV strains were clustered in subclade A1 with the other FWPV strains. They showed an average of 99.87, 87, 89.39, and 88.24 % nucleotide identity to the other strains in subclade A1, A2, A3, and A4, respectively, and 66.67 % nucleotide identity to CNPV-like group of viruses (Clade B). Elsharqyia_PGPV strain was clustered in subclade A4 together with the other PGPVs (PGPVP and PGPVIndia_08_2009). Hence, it showed an average of 87.58, 94.8, 97, and 98.7 % nucleotide identity to subclade A1, A2, A3, and A4, respectively, and 66.50 % nucleotide identity to the CNPV-like group of viruses (Clade B) (Fig. 4).

Phylogenetic analysis of APVs based on fpv140 (H3L) gene nucleotide sequences. Phylogenetic tree was constructed via multiple alignments of 892-bp nucleotide sequence of H3L gene from 22 APV strains. The tree was analyzed by neighbor-joining (N-J) analysis with bootstrapping (1000). APV subclades A1, A2, A3, A4, and B1 are labeled. (Asterisk) Elsharqyia_FWPV represents Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, and Elsharqyia_FWPV4. See Tables 1 and 2 for virus abbreviations
Discussion
Most of our knowledge about the situation of APVs in Egypt was obtained from a very few reports [2, 7, 17] concerning the epidemiology and the infection biology of the virus. The incidence of APV infection has been recorded before among different avian species in the Elsharqyia Province in a period of 2 years (Late summer 1996–Spring 1998) [6]. However, and up till now, there was no available record of the molecular composition of APVs circulating in Egypt. In this study, a small scale survey was conducted to understand the nature of APVs isolated from different backyard domesticated birds in the Elsharqyia province (Egypt). The samples were collected during the summertime in 2011 as it is the prime mosquito season in Egypt [19, 22]. Six APVs were isolated from chickens, turkeys, and pigeons on CAM of ECE. For most of the samples, the pocks were clear after the first passage suggesting their ability for embryonic adaption as previously reported [13]. The evidence of virus growth was confirmed by the histopathologic examination of Pox virus lesion on CAMs. Moreover, the isolated viruses were molecular biologically analyzed and characterized. As previously shown, PCR amplification of fpv140 and fpv167 gene loci were used in many reports for the molecular identification and characterization of the APV genome in a different era all over the world [11–13, 20]. Consistently, in our studies, PCR amplification of the P4b (fpv167) gene revealed the highly conserved 578-bp APV-specific DNA fragment. Based on the length of the amplicons, the amplification of fpv140 locus allows the discrimination between FWPV and CNPV [11]. Since the PCR of fpv140 gene locus revealed 1,800-bp PCR products, all the studied isolates in the present study were classified as FWPV or FWPV-like viruses [11]. This result in contrast to the previous finding [13], as it was suggested the inapplicability of fpv140 gene as a diagnostic marker for APVs. Sequencing of the P4b-578-bp DNA fragment was performed on six isolates from different host species. The sequencing results revealed that all the isolates belonged to the major clade A according to [11]. Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV showed 100 % identity and clustered in the minor subclade A1 (FWPVs) with the two commercially available vaccines FWPVN and FWPVFPV9, as well as the other poxviruses detected from chickens, turkeys, and sparrows. It is not obvious if the clustering of the Elsharqyia_TKPV with the FWPVs in the same subclade is due to the emergence of FWPVs to the turkey host or both viruses are closely related [1, 12]. The five strains, Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV, revealed mean distance percentage of 28 and 29 % with clade B and C, respectively.
As previously mentioned by Jarmin et al. [11], PGPV would not be used as an independent taxonomic unit based on the phylogeny of fpv167 (P4b) gene as viruses from clades A and B can infect and cause disease in pigeons. Elsharqyia_PGPV was clustered in the minor subclade A2 together with TKPVs, PEPVs, OSPVs, and other PGPVs. Hence, it was the nearest phylogenetic neighbor and showed 100 % nucleic acid identity with the wood pigeon Indian isolate (PGPVIndia_08_2009) and the unknown origin pigeon isolates (PGPVP & PGPVTP-2) [11, 12]. In addition, Elsharqyia_PGPV revealed the mean distance percentage of 28 and 30 % with clade B and C, respectively.
The phylogenetic analysis of the fpv140 gene allows easily differentiation between the different viruses: subclades A1 (FWPV), A2 (TKPV), A3 (FLPV), A4 (PGPV), and B1 (CNPV) [5, 11, 15]. Consistently, in our study, the phylogenetic analysis of the fpv140 from the Egyptian APVs clustered the five viruses isolated from chicken and the one from turkey in subclade A1 (FWPV). The five strains revealed a distance percentage of 56.9 % from clade B. On the other hand, it places Elsharqyia_PGPV with the other PGPVs in the subclade A4 with a distance percentage of 53.9 % from clade B. Our study also supports the previous finding [11, 15] that the phylogenetic analysis of APVs based on the fpv140-H3l gene allows clustering of the PGPV strains in an independent taxonomic unit which correlates with the pigeon host species.
In conclusion, sequence analysis of the APV-fpv167 and APV-fpv140 revealed that Elsharqyia_FWPV1, Elsharqyia_FWPV2, Elsharqyia_FWPV3, Elsharqyia_FWPV4, and Elsharqyia_TKPV belong to Fowlpox-like viruses (subclade A1). None of the analyzed strains belong to either the Canarypox-like virus isolates (Clade B) or Psittacinepox-like virus isolates (Clade C). The phylogenetic analysis of the fpv140 allows better discrimination for Elsharqyia-PGPV (subclade A4) from the FWPV strains. Taken together, this small scale study provides the first molecular characterization of Egyptian APVs. To have a better understanding about the genetic composition of APVs and their relationship to each other and to their host species a larger scale survey needs to be performed. Hence, it should include a wider range of hosts and kinds of management system to study the evolution of APVs in Egypt.
Materials and methods
Viruses
Six isolates of APV from different avian species were used in this study. These viruses were isolated from chickens, turkeys, and pigeons which were reared in the backyard management system of some villages in Elsharqyia province, Egypt. These birds were suffering from the skin lesions of APV infection on the comb, eyelids, beak, and wattles as well as the diphtheritic membrane on the mucous membrane of the upper respiratory tract. The affected flocks suffered from low or even no mortality except in case of pigeon; hence, there was a high level of mortality among the squabs reach to 30 %. Commercially available live-vaccinal strain against FWPV (FWPVFPV9) was also included in the study as shown in Table 1.
Virus isolation in embryonated chicken eggs (ECE)
Scab samples from each clinical case were minced separately using sterile iced mortar and pestle, suspended in phosphate buffer saline containing 1,000 IU/ml penicillin, and 100 μg/ml streptomycin. Sample suspensions were centrifuged at 2,000 rpm/10 min, thereafter 100 μl from each sample supernatant was separately inoculated onto chorioallantoic membrane (CAM) of 13-day-old ECE from non-vaccinated flock. The inoculated eggs were incubated at 37 °C for 5 days and then examined for focal white pock lesions or generalized thickening of the CAMs. Portions of CAM showing lesions were frozen at-20 °C and were used for DNA extraction.
Histopathology
Both skin and pock lesions were fixed in 10 % buffered formalin and embedded in paraffin. Thin sections of the fixed lesions were stained with hematoxylin and eosin stain and examined microscopically for the presence of intracytoplasmic inclusion bodies.
DNA extraction
DNA was extracted from 20 mg of frozen cutaneous lesion and CAMs as well as 20 mg of lyophilized vaccine using GeneJET™ genomic DNA Purification Kit (Fermentas) following the manufacturer’s instructions.
PCR amplification
PCR for amplification APVs specific fragments (fpv167 and fpv140 genes) were performed. Primers described previously [10, 13] were used to amplify the FWPV P4b gene (fpv167 locus) (Forward primer: 5′-CAGCAGGTGCTAAACAACAA-3′ & reverse primer: 5′-CGGTAGCTTAACGCCGAATA-3′) as well as fpv140 gene (Forward primer: 5′-GAAGTAGAGTTATCGGTTC-3′ & reverse primer: 5′-GGTGATCCATTTCCATTTC-3′). Amplification was performed in an Eppendorf thermal cycler (MWG) under the following conditions: cycle at 95 °C for 2 min (initial denaturation), 40 cycles (denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 1 min), and cycle of final extension at 72 °C for 10 min. Negative controls were included in each assay for detection of any contamination using Dream Taq™ Green PCR Master Mix (2×) (Fermentas). Five microliters of amplified PCR products were separated by 1 % ethidium bromide strained agarose gel electrophoresis at 120 V for 20 min. 1-kbp DNA Marker (Fermentas) was used as standard and the amplified products were visualized using ultraviolet light transilluminator (Spectroline).
Purification and sequencing of PCR product
PCR products were purified using Gene JET PCR purification kit (Fermentas) and resuspended in 50 μl H2O. Each purified amplicon was sequenced in both forward and reverse directions using the amplification primers. The sequencing reaction was performed in an automated sequencer (Macrogen Inc., Korea ABI 3730XL DNA analyzer). The sequences were submitted to the Genbank database under accession numbers which were listed in Table 1.
Phylogenetic analysis
Comparative alignment and phylogenetic analyses were performed computationally by means of CLC main workbench program. The phylogenetic tree was generated by N-J tree method and the liability of internal branches was assessed by 1000 bootstrap replications.
References
C.L. Afonso, E.R. Tulman, Z. Lu, L. Zsak, G.F. Kutish, D.L. Rock, The genome of fowlpox virus. J. Virol. 74, 3815–3831 (2000)
M.M. Amer, A.M.W. Kheir Eldin, M.H.H. Awaad, Studies on pox in Japanese quail (Coturnix Coturnix Japonica). J. Egypt. Vet. Med. Assoc. 46, 295–302 (1986)
S.K. Biswas, C. Jana, K. Chand, W. Rehman, B. Mondal, Detection of fowl poxvirus integrated with reticuloendotheliosis virus sequences from an outbreak in backyard chickens in India. Vet. Ital. 47, 147–153 (2011)
A. Bolte, J. Meurer, E. Kaleta, Avian host spectrum of avipoxviruses. Avian Pathol. 28, 415–432 (1999)
O. Carulei, N. Douglass, A.L. Williamson, Phylogenetic analysis of three genes of Penguinpox virus corresponding to Vaccinia virus G8R (VLTF-1), A3L (P4b) and H3L reveals that it is most closely related to Turkeypox virus, Ostrichpox virus and Pigeonpox virus. Virol. J. 6, 52 (2009)
A.A.M. Eid, M.A. EL-Said, I.A. Ghanem, Pox Virus Infection Among Some Avian Species In Sharkia Province (4th Veterinary Medicine Zagazig Congress, Hurghada, 1998), pp. 681–688
K.S. EL-Zanaty, Identification and characterization of pox virus isolated from turkey. Assiut Vet. Med. J. 23, 240–248 (1990)
C. Gubser, S. Hue, P. Kellam, G.L. Smith, Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 85, 105–117 (2004)
H.J. Ha, L. Howe, M. Alley, B. Gartrell, The phylogenetic analysis of avipoxvirus in New Zealand. Vet. Microbiol. 150, 80–87 (2011)
L. Huw Lee, K. Hwa Lee, Application of the polymerase chain reaction for the diagnosis of fowl poxvirus infection. J. Virol. Methods 63, 113–119 (1997)
S. Jarmin, R. Manvell, R.E. Gough, S.M. Laidlaw, M.A. Skinner, Avipoxvirus phylogenetics: identification of a PCR length polymorphism that discriminates between the two major clades. J. Gen. Virol. 87, 2191–2201 (2006)
D. Luschow, T. Hoffmann, H.M. Hafez, Differentiation of avian poxvirus strains on the basis of nucleotide sequences of 4b gene fragment. Avian Dis. 48, 453–462 (2004)
G. Manarolla, G. Pisoni, G. Sironi, T. Rampin, Molecular biological characterization of avian poxvirus strains isolated from different avian species. Vet. Microbiol. 140, 1–8 (2009)
M. Mohan, T.F. Fernandez, A case report of Pigeon pox-histopathologic diagnosis. Vet. World 1, 117–118 (2008)
R.M. Pawar, S.S. Bhushan, A. Poornachandar, U. Lakshmikantan, S. Shivaji, Avian pox infection in different wild birds in India. Eur. J. Wildl. Res. 57, 785–793 (2011)
J. Perez-Tris, R.A. Williams, E. Abel-Fernandez, J. Barreiro, J.J. Conesa, J. Figuerola, M. Martinez–Martinez, A. Ramirez, L. Benitez, A multiplex PCR for detection of poxvirus and papillomavirus in cutaneous warts from live birds and museum skins. Avian Dis. 55, 545–553 (2011)
M. Saif-Edin, S.S. El-Ballal, Epidemiological and ultrastructural studies on Pigeon pox in upper Egypt. Assiut Vet. Med. J. 37, 68–85 (1997)
H.L. Shivaprasad, T. Kim, D. Tripathy, P.R. Woolcock, F. Uzal, Unusual pathology of canary poxvirus infection associated with high mortality in young and adult breeder canaries (Serinus canaria). Avian Pathol. 38, 311–316 (2009)
D.N. Tripathy, W.M. Reed, Pox, in Diseases of Poultry, 11th edn., ed. by Y.M. Saif, H.J. Barnes, J.R. Glisson, A.M. Fadly, L.R. McDougald, D.E. Swayne (Iowa State University Press, Ames, 2003), pp. 253–269
S.C. Weli, M.I. Okeke, M. Tryland, O. Nilssen, T. Traavik, Characterization of avipoxviruses from wild birds in Norway. Can. J. Vet. Res. 68, 140–145 (2004)
S.C. Weli, T. Traavik, M. Tryland, D.H. Coucheron, O. Nilssen, Analysis and comparison of the 4b core protein gene of avipoxviruses from wild birds: evidence for interspecies spatial phylogenetic variation. Arch. Virol. 149, 2035–2046 (2004)
S.C. Weli, M. Tryland, Avipoxviruses: infection biology and their use as vaccine vectors. Virol. J. 8, 49 (2011)
Acknowledgments
We are thankful to Prof Dr Abd El-Moneim Ahmed Ali, Dr Haitham Alyadin, and Mostafa Hassan (Zagazig University) for histopathologic examination and imaging. Ahmed El-Baqer for PGPV clinical sample provision.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Abdallah, F.M., Hassanin, O. Detection and molecular characterization of avipoxviruses isolated from different avian species in Egypt. Virus Genes 46, 63–70 (2013). https://doi.org/10.1007/s11262-012-0821-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0821-y