
Abstract
Buffalopox virus (BPXV), a close variant of vaccinia virus (VACV) has emerged as a zoonotic pathogen. The host tropism of poxviruses is governed by host-range genes. Among the host-range genes: E3L, K3L, and C7L are essential for virus replication by preventing interferon resistance, whereas B5R is essential for spread of the virus and evasion from the host’s immune response as in VACV. We report sequence analysis of host-range genes: E3L, K3L, C7L, and membrane protein gene (B5R) of BPXVs from buffalo, cattle, and human from recent outbreaks in India—their phylogenetic relationship with reference strain (BP4) and other Orthopoxviruses. BPXVs revealed a sequence homology with VACVs including zoonotic Brazilian VACV-like viruses. The aa sequences of E3L and K3L genes were 100 % similar in buffalo, cattle, and human isolates. However, four significant point mutations (I11K; N12K and S36F in C7L gene and D249G in B5R gene) were observed specific to buffalo isolate only. This signifies that different strains of BPXV were circulated during the outbreak. The mutations in C7L and B5R could play an important role in adaptation of BPXV in human and cattle which needs further functional studies. The strain of BPXV isolated from buffalo may not be adopted in human and cow. Various point mutations were observed in the host-range genes of reference strain (BPXV-BP4) which may be due to several passages of virus in cell culture. The phylogeny constructed based on concatenated gene sequences revealed that BPXVs are not as closely related to vaccine strain (Lister and Lister-derived strain—LC16m8), as hypothesized earlier, rather they are more closely related to reference strain (BPXV-BP4) and other vaccinia and vaccinia-like viruses such as Passatempo and Aracatuba viruses. The availability of information regarding host tropism determinants would allow us to understand molecular mechanism of species tropism of poxviruses which would be useful in unveiling new strategies to control zoonotic poxviral infections.
Similar content being viewed by others


Introduction
Buffalopox is an emerging contagious viral zoonosis of domestic buffaloes (Bubalus bubalis) often associated with high morbidity (80 %) in affected herds, infecting milkers [1] and cows [2]. The disease is characterized by localized pock-lesions on the udder, teats, inside of thighs, base of the ears, inner surface of earflap and eyes [3]. The causative agent of the disease, BPXV, is a close variant of vaccinia virus which is a type-species of the genus Orthopoxvirus (OPXV) under Poxviridae family [4]. The WHO Joint Expert Committee on Zoonosis [5] declared buffalopox as an important zoonotic disease.
It has been speculated that BPXV has emerged from the vaccine strain (Lister strain) of vaccinia virus which was used to produce smallpox vaccine in buffalo calves in India [6]. Gradually, the vaccine strain adapted in buffaloes and became pathogenic causing outbreaks in buffaloes. Thereafter, BPXV outbreaks have been frequently occurring in different parts of the country affecting buffaloes [1, 7–9] and humans [1, 6, 10–13]. Zoonotic cases of BPXV have also been reported from Pakistan [14]. Subsequently, cases of BPXV in spillover hosts like cows have also been reported [2]. Likewise, vaccine strain-derived pathogenic vaccinia-like viruses viz., Cantagalo [15], Aracatuba [16], and Passatempo [17] viruses are being reported from Brazil. Further, transmission of bovine vaccinia virus to humans has also been reported in Brazil [18–20]. Transmission of the virus to cows and humans indicates change in host-specificity which could have serious implications from a public health perspective due to the cessation of vaccination against smallpox since 1977 [21]. The studies on host-range genes and genes involved in morphogenesis of virus could have a bearing on adaptation of the virus to spillover hosts thereby elucidating their role in host tropism.
The product of host-range genes have been shown to influence the ability of the virus to infect cells by subverting the host immune response [22]. The most important host-range genes identified in VACV are E3L, K3L, and C7L [22] which have been implicated in alteration of host cell antiviral defense mechanism. The E3L gene encodes a 20- and 25-kDa protein that represses the host cell antiviral response by inhibition of both protein kinase and RNaseL [23–26]. While K3L gene confers interferon resistance [27, 28] and is shown to suppress protein kinase (PKR) activation and eIF2α phosphorylation in mammalian cells [29] which could lead to inhibition of antiviral defense mechanism. The C7L gene is conserved in all the Orthopoxvirus genomes [30] and causes inhibition of apoptosis [31] and blocking of antiviral effects by antagonizing interferons (IFN) [32]. The B5R gene is essential for formation of extracellular enveloped virus (EEV) [33] and involved in viral evasion from the host’s immune response [34]. The B5R protein has conserved extracellular domain homologous to complement regulatory proteins, which may be involved in viral evasion from host immune responses by protecting complement-mediated attack [35]. Deletion of this gene of VACV strain—WR decreases the production of EEV, reduced plaque size in vitro and is highly attenuated in vivo compared to the parental strain [36]. Keeping the above facts in view this study envisages to determine the full-length sequences of the E3L, K3L, C7L, and B5R genes of BPXVs isolated from outbreaks in buffaloes, humans, and cattle in India, and elucidate the evolutionary relationship with other OPXVs circulating in the world vis-a-vis the reference strain.
Materials and methods
Viruses
The BPXVs isolated from recent outbreaks (2010 and 2011) in buffaloes (BPXV/buffalo/Jalgaon/2010) and humans (BPXV/human/Jalgaon/2010) from Maharashtra and cattle (BPXV/cow/Baatnor/2011) from Uttar Pradesh states of India were utilized in the study. The viruses were isolated from scabs in Vero cell culture in two to three passages. The reference strain (BP4) of BPXV available in the VTCC repository was utilized for comparison purpose. The BPXV-BP4 strain was originally isolated from outbreak in buffaloes in Hisar, Haryana, India at the Department of Bacteriology and Hygiene, Haryana Agricultural University, Hisar (India) [37].
Isolation of viral DNA
Viral DNA was extracted from 200 μl of infected Vero cell culture fluid using the AuPreP™ DNA Extraction Kit (Life Technologies Pvt. Ltd., New Delhi, India) as per the manufacturer’s protocol. The DNA was eluted in 50 μl nuclease-free water and an aliquot of 5 μl was used for PCR amplification.
Polymerase chain reaction, cloning, and sequencing
Full-length sequence of E3L, K3L, C7L, and B5R genes were amplified using gene-specific primers commercially synthesized from Sigma-Aldrich, USA. The primer sets were designed on the basis of genome sequence of VACV-WR strain (Accession no. AY243312) using Primer-BLAST software (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The following primer sets were used to amplify the complete coding sequences of the targeted genes: Forward-5′-ACGACGAACCACCAGAGGATGATGA-3′ and Reverse-5′-TCATCTTTAGAGAATATACTAGTCGCGTT-3′ for E3L gene; Forward-5′-TCCCAATTTACGAGCC CGTTAACAA-3′ and Reverse-5′-TTTTATGTTCGGTATAAAAATTATTGATGTCTAC-3′ for K3L gene; Forward-5′-ATGGGTATACAGCACGAATTCGACATCATT-3′ and Reverse-5′-TTAATCCATG GACTCATAATCTCTATACGGG-3′ for C7L; and Forward-5′-TTTCCTTTTAGTGCTCGACAGTGTA-3′ and Reverse-5′-AACGGATTTATATTTACGG TAGCAA-3′ for B5R gene. PCR reaction was set up using 5 U of DreamTaq™ DNA polymerase (MBI Fermentas, Burlington, Canada). The PCR was performed with the thermal cycling condition of 95 °C for 5 min followed by 35 cycles of 95 °C for 1 min, 51 °C for 50 Sec, 72 °C for 50 s, and final extension at 72 °C for 10 min. The amplified products were purified using PCR purification kit (Stratagene, La Jolla, CA, USA) as per the manufacturer’s protocol and cloned into pTZ57R/T vector (MBI Fermentas, Burlington, Canada) following standard procedure. The recombinant plasmid DNA was confirmed by colony PCR, restriction enzyme analysis, and sequencing. All clones were sequenced commercially employing automated DNA sequencer (ABI 3130 Genetic Analyzer) by Chromas Biotech Asia Pvt. Ltd., Bangalore, India. Three clones for each gene were selected for sequencing and a consensus sequence was obtained for analysis.
Sequence and phylogenetic analysis
An open reading frame (ORF) nucleotide homology search was carried out using NCBI BLAST server [38]. For comparison, nucleotide (nt) as well as deduced amino acid (aa) sequences were further aligned using Clustal W method of molecular evolutionary genetic analysis (MEGA) version 5 program [39]. Gene sequences of all the members of the OPXV genus were retrieved from the NCBI database (http://www.ncbi.nlm.nih.gov/). The details of these sequences including their accession numbers are given in Table 1. The sequence identity was determined at the nucleotide and amino acid sequence levels. Hydrophathicity values of the each amino acid were calculated using the ProtScale tool at ExPASy server (http://www.expasy.org/tools/protscale.html) choosing the hydrophobicity scale of Kyte & Doolittle with window size 19.
For building the phylogenetic tree, aligned individual gene sequences were manually edited and concatenated together. A Maximum Likelihood rooted phylogenetic tree was constructed from the artificially concatenated ORF sequences using MEGA 5 [39] and the tree topologies were evaluated by bootstrap using 1,000 replicates of the data set.
Results
Amplification and sequencing of genes: E3L, K3L, C7L, and B5R
Full-length E3L, K3L, C7L, and B5R genes of (BPXV/buffalo/Jalgaon/2010), (BPXV/human/Jalgaon/2010), and (BPXV/cow/Baatnor/2011) were successfully amplified using gene-specific primers, cloned, and sequenced. First three genes of reference strain (BP4) were also amplified and sequenced as the sequence of B5R gene is available in the database. The amplicons sizes were 787, 313, 453, and 1,019 bp for E3L, K3L, C7L, and B5R genes, respectively. The nt sequences were submitted to GenBank and assigned accession numbers (JN653080, JN653079, JN653081, and JN653082 for E3L gene; JN653084, JN653083, JN653085, and JN653086 for K3L gene; JN653088, JN653087, JN65308, and JN653090 for C7L gene and JN653092, JN653091, and JN653093 for B5R gene).
Homology analysis
The ORF of the targeted host-range genes of all three BPXVs comprised of 573, 267, 453, and 954 bp nucleotide encoding predicted 190, 88, 150, and 317 aa proteins for E3L, K3L, C7L, and B5R, respectively. Comparison of sequences revealed that E3L and K3L genes of buffalo isolate (BPXV/buffalo/Jalgaon/2010) shared 100 % similarity with cattle (BPXV/cow/Baatnor/2011) and human (BPXV/human/Jalgaon/2010) isolates at both nt and aa levels, as compared to C7L and B5R genes which showed a similarity of 96.67–99.58 %. The reference strain (BP4) revealed a similarity of 92.63–98.88 % at both nt and aa levels for E3L, K3L, and C7L genes in comparison to BPXVs of this study; whereas B5R gene had a comparatively higher similarity of 99.48 % at nt and 99.05 % at aa level. The BPXV isolates revealed a higher degree (97.84–99.65 % at nt and 95.06–99.43 % at aa level for E3L; 98.50–99.63 % at nt and 97.73–100 % at aa level for K3L; 98.01–98.68 % at nt and 97.33 % at aa level for C7L; and 97.80–99.69 % at nt and 96.85–99.05 % at aa level for B5R genes) of similarity with VACV.
Analysis of amino acid changes
The detailed amino acid changes in the encoded protein sequences have been depicted in Tables 2, 3, 4, and 5 for E3L, K3L, C7L, and B5R proteins, respectively. Analysis of E3L protein revealed three consensus aa changes (N21T; A79V and V85A) at N-terminal region, whereas two aa changes (I123V and K132R) were noted at the C-terminal region of the Indian BPXV isolates (Table 2). However, aa changes (N21T and I123V) also observed in VACV-Lister strain. These aa changes did not affect the hydropathic properties of the amino acids. Further, sixteen sense mutations (A55T, T163C, A253T, A255C, G256A, T261G, G288T, A295C, G304C, A305T, A308T, T311C, T329C, A335C, A349C, and T376C) leads to fourteen mutational changes in this protein of reference strain (BPXV-BP4) (Table 2). The evaluation of the K3L gene sequences did not reveal any significant changes compared to VACV, however, three aa changes (S7L, A38D, and C85R) were observed in K3L protein of reference strain (BP4) (Table 3). Four significant point mutations (I11K; N12K, S36F, and F108L) were found in C7L protein of the buffalo isolate (BPXV/buffalo/Jalgaon/2010) compared to other OPXVs including VACV and VARV (Table 4). These changes were not observed in cattle and human BPXV isolates except point mutation (F108L) in human isolate. Amino acid changes (I11K and N12K) led to significant decrease in hydrophobicity, whereas, aa change (S36F) causes change in hydrophilicity. However, the reference strain (BP4) showed seven aa changes in this protein sequence (Table 4). Sequence analysis of B5R proteins showed several aa changes in Indian isolates (Table 5). We observed three point mutations each in buffalo (D64N; S240F and D249G) and cattle (F46L; E111K and S197P) isolates; and four mutations in human (D64N; F215L; S/T240F and F292L) isolate in comparison to OPXVs. However, aa residue (D) in human and cattle isolates observed at position 249 along with other OPXVs including Brazilian VACV strains (Table 5) which caused little increase in hydrophilicity property.
Phylogenetic analysis
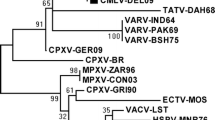
The topology of the rooted phylogenetic tree of the concatenated sequences of four genes revealed that BPXVs of present outbreaks were closely related to the reference strain (BPXV-BP4) with bootstrap value of 94 (Fig. 1). Broadly, BPXVs showed grouping with bootstrap value-79 with VACV strains—Duke, Ankara, Modified Ankara virus, and Copenhagen, Western Reserve along with recently reported vaccinia-like viruses viz., Passatempo and Aracatuba viruses. However, vaccine strains (VACV-Lister and VACV-LC16m8, derived strain from Lister) grouped separately.

Maximum-likelihood tree constructed using concatenated alignment of nucleotide sequences of E3L, K3L, C7L, and B5R genes of BPXV isolates reported in this manuscript and previously reported sequences of OPXVs using MEGA5. The bootstrap scores (1,000 replicates) for all nodes are displayed next to the branches. The representative sequences reported in this manuscript are marked by black squares
Discussion
The host tropism of the poxvirus is regulated by host-range genes which are responsible for modulation of intracellular events after binding and entry of the virus into the host cells [22]. The detailed study of host-range genes of poxviruses is a prime concern worldwide in the light of changes in host-specificity of the VACV, BPXV, and other members of genus Orthopoxvirus to spillover hosts including humans. Further, the world population is naïve to the poxviruses due to cessation of smallpox vaccination in 1977 [21] which could have serious public health implications. Further, genes responsible for formation of enveloped virus play critical role in cell-to-cell and long-range virus spread. In this study, analysis of the selected host-range genes viz., E3L, K3L, C7L genes and gene involved in morphogenesis of virus viz., B5R gene of BPXV isolates of buffalo, human, and cattle origin could pave the way toward finding a solution to the underlying cause of zoonosis and host tropism of poxviruses despite their strict species-specificity [22]. Further, a comparative sequence analysis was carried out to understand the genetic relationship of BPXVs of different species origin and to other OPXVs and to explore variations in these genes. The sequences of the three host-range genes and one structural gene were also compared with the reference strain (BPXV-BP4) to determine changes in recent isolates.
All BPXV isolates from buffalo, human, and cattle were used to carry out the genetic analysis of the host-range and structural genes. All the targeted genes were successfully amplified by PCR using template DNA extracted from virus. Comparison of sequences revealed that E3L and K3L genes were 100 % similar among buffalo, cattle and human isolates at both nt and aa levels, whereas C7L and B5R genes showed a similarity of 96.67–99.58 %. Further, BPXV isolates showed a higher degree (97.84–99.65 % at nt and 95.06–99.43 % at aa level for E3L; 98.50–99.63 % at nt and 97.73–100 % at aa level for K3L; 98.01–98.68 % at nt and 97.33 % at aa level for C7L; and 97.80–99.69 % at nt and 96.85–99.05 % at aa level for B5R genes) of similarity with VACV. These indicate that the studied genes were highly conserved among Indian isolates and other OPXVs. However, the reference strain (BP4) revealed a lower percentage of similarity (92.63–98.88 % at both nt and aa levels) for E3L, K3L, and C7L genes in comparison to all BPXV isolates; however, B5R gene showed similarity of 99.48 % at nt and 99.05 % at aa level.
Amino acid sequence analysis revealed consensus aa changes at N-terminal (N21T; A79V and V85A) and C-terminal regions (I123V and K132R) of E3L protein of all BPXV isolates (Table 2) without any change in hydropathic properties. However, several sense mutations in nt sequence of this gene of BPXV-BP4 reference strain leads to fourteen aa changes which indicate the loss of functional activities and virulence. Mutations (N22K and S36F) observed in K3L proteins in recent zoonotic Brazilian VACV-like viruses viz, Aracatuba and Passatempo were absent in BPXVs and these mutations did not cause major change in hydropathic properties. Earlier mutational studies of His47Gln, His47Asp, Val52Lys, and Val44Gln sites indicated that these mutations perturb the maximum efficiency of PKR inhibitory function of K3L protein [40–42]. The presence of all these sites in VACV and BPXVs indicates the highly conserved nature of the gene and its functional significance in host pathogenesis. However, in reference strain (BPXV-BP4) three aa changes (S7L, A38D, and C85R) were observed. The C7L protein regulates cellular tropism of VACV, and is highly conserved among Orthopoxviruses [22, 43]. Interestingly, three amino acid residues (Lys, Lys, and Phe) were observed at position 11, 12, and 36 in BPXV strain isolated from buffalo, whereas, Ile, Asn, and Ser amino acid residues were found in human and cattle isolates along with other OPXVs including VACV and VARV (Table 4). This change of basic aa (Lysine) to neutral aa (Isoleucine/Asparagine) and increased hydrophobicity may lead to structural and functional activity of the protein. Similarly, in B5R protein, the amino acid residue—Glycine is present at position 249 in buffalo isolate, whereas, Aspartic acid present in both human and cattle isolates along with all OPXVs including reference strain (BPXV-BP4) and Brazilian VACV strains. Further, this gene is not related to high passage number as minimal changes were observed in reference strain (BP4) which had undergone several passages in cell culture in the past (>50). This indicates that either different strains of BPXV were circulating in the outbreak as has been reported in Brazil where two strains of vaccinia virus were found to be circulating in a single outbreak of bovine vaccinia virus [44] or mutation could have taken place in human and cattle isolates which could be critical for adaptation of BPXV in these spillover hosts. However, BPXV strain circulating in buffaloes may not be adaptable in human and cattle as the mutations in BPXV/buffalo/Jalgaon/2010 strain were exclusively confined to buffaloes as compared to other OPXVs. The structural and functional studies on unique mutations observed in C7L and B5R proteins is required to elucidate their role in host tropism of BPXV.
To evaluate the evolutionary relationship of BPXV isolates with OPXVs, a concatenated phylogenetic tree was constructed considering all genes together to produce robust tree supported by high-bootstrap values. This approach is suitable for vaccinia viruses because the evolution of these viruses taking place in isolation and further there’s been no evidence of recombination of genomes of vaccinia viruses which could have an effect on analyses. The phylogram of the concatenated gene sequences showed clustering of BPXVs with VACVs (Fig. 1) with high bootstrap value (79 %) which indicates that these genes are conserved among VACVs. Interestingly, VACV vaccine strain (Lister) and LC16m8, derived strains from Lister clustered separately which indicates that these two strains could be probable ancestors of BPXV and other VACV strains. However, phylogeny revealed that BPXVs are closely related to Duke strain of VACV. In this regard, the ancestry of Duke strain was traced back to the American New York Board of Health strain, isolated from vaccine-related complication of a female patient vaccinated with smallpox vaccine-Dryvax [45–47], thereby contradicting our hypothesis stating BPXVs in India have basically originated from vaccine strain (Lister) that were mainly produced in buffalo calves. Hence, it can be hypothesized that the BPXV might have evolved through gradual adaptation of the vaccine strain in buffaloes. In this context, the analysis of whole genome sequence data could be useful in validating this hypothesis. The grouping of zoonotic Brazilian VACV-like viruses (Aracatuba and Passatempo viruses) with VACVs and BPXVs indicates that in India only BPXV is circulating among buffalo, human, and cattle, as there are no reports of Cowpox in the country off late [1, 2, 7]. The analysis of the phylogram indicates that the host-range and structural genes are highly conserved among Orthopoxvirus genus and play a critical role in pathogenesis.
To conclude, this is the first report on genetic characterization of important host-range genes of BPXV isolates of buffalo, cattle, and human origin except for the B5R gene of buffalo isolate which was reported earlier [48]. The sequence and phylogenetic analysis revealed that recent BPXVs are closely related to reference strain (BPXV-BP4) and VACV which corroborates the previous findings [7, 48, 49]. In contrast to robustness of close clustering of the reference strain (BPXV-BP4) and recent BPXVs supported by high bootstrap value (94 %), there were several mutational changes in the individual host-range genes. This is due to several passages of BP4 strain in cell culture in the past (>50) to develop as live attenuated vaccine. Furthermore, phylogeny revealed that BPXVs are not as closely related to vaccine strain (Lister and Lister-derived strain—LC16m8), as hypothesized earlier, rather they are more closely related to Indian vaccine strain (BPXV-BP4) and other vaccinia and vaccinia-like viruses such as Passatempo and Aracatuba viruses. However, analysis of whole genome sequence data may provide insight to confirm this hypothesis conclusively. A better understanding of molecular mechanisms that govern the species tropism of poxvirus in non-evolutionary hosts is essential. The unique portfolio of immunomodulatory and host-range genes governs the poxvirus tropism at cellular level and this, in turn, imparts unique properties of host-range, pathogenesis and potential for host-to-host spread in each poxvirus. The genetic information of the important host-range and structural genes of BPXVs of buffalo, cattle, and human would be useful in understanding their pathogenesis in spillover host so as to devise strategies for the control of the zoonosis.
References
R.K. Singh, M. Hosamani, V. Balamurugan, V. Bhanuprakash, T.J. Rasool, M.P. Yadav, Anim. Health Res. Rev. 8, 105–114 (2007)
S. Yadav, M. Hosamani, V. Balamurugan, V. Bhanuprakash, R.K. Singh, Arch. Virol. 155(2), 255–261 (2010)
K.P. Mallick, Indian J. Vet. Med. 8, 146–147 (1988)
F.A. Murphy, E.P.J. Gibbs, M.C. Horzinek, M.J. Studdert, in Veterinary Virology, 3rd edn. (Academic Press, San Diego, 1999)
FAO/WHO, Third Report, WHO Technical Report Series, Geneva, (1967), p. 378
R.K. Singh, M. Hosamani, V. Balamurugan, C. Satheesh, T.J. Rasool, M.P. Yadav, Arch. Virol. 151, 1995–2005 (2006)
R.K. Singh, M. Hosamani, V. Balamurugan, C.C. Satheesh, K.R. Shingal, S.B. Tatwarti, R.G. Bambal, V. Ramteke, M.P. Yadav, Sci. Tech. Rev. O. I. E. 25, 981–987 (2006)
S.M. Lal, I.P. Singh, Trop. Anim. Health Prod. 9, 107–112 (1977)
R. Chandra, S.K. Garg, U.V.S. Rana, V.D.P. Rao, Farm Anim. 2, 57–59 (1987)
S. Nedunchelliyan, D.S. Reddy, K.S. Venkataraman, Indian J. Public Health 24, 2 (1992)
R.M. Kolhapure, R.P. Deolankar, C.D. Tupe, C.G. Raut, A. Basu, B.M. Dama, S.D. Pawar, M.V. Joshi, V.S. Padbidri, M.K. Goverdhan, K. Banerjee, Indian J. Med. Res. 106, 441–446 (1997)
V. Bhanuprakash, G. Venkatesan, V. Balamurugan, M. Hosamani, R. Yogisharadhya, P Gandhale, K.V. Reddy, A.S. Damle, H.N. Kher, B.S. Chandel, H.C. Chauhan, R.K. Singh, Zoonoses Public Health (2010). doi:10.1111/j.1863-2378.2009.01314.x
Y.K. Gurav, C.G. Raut, P.D. Yadav, B.V. Tandale, A. Sivaram, M.D. Pore, A. Basu, D.T. Mourya, C.M. Akhilesh, Prev. Vet. Med. 100, 242–247 (2011)
A. Zafar, R. Swanepoel, R. Hewson, M. Nizam, A. Ahmed, A. Husain, A. Grobbelaar, K. Bewley, V. Mioulet, B. Dowsett, L. Easterbrook, R. Hasan, Emerg. Infect. Dis. 13(6), 902–904 (2007)
C.R.A. Damaso, J.J. Espostio, R.C. Condit, N. Moussatche, Virology 277, 439–449 (2000)
G.S. Trindadae, F.G. Fonseca, J.T. Marques, M. Nogueira, L.C. Mendes, A.S. Borges, E.M. Pituco, C.A. Bonjardim, P.C. Farreira, E.G. Kroon, Emerg. Infect. Dis. 9, 155–160 (2003)
J.A. Leite, B.P. Drumond, G.S. Trindade, Z.I.P. Lobato, F.G. da Fonseca, J.R. Sandos, M.C. Madureira, M.I. Guedes, J.M. Ferreira, C.A. Bonjardim, P.C. Ferreira, E.G. Kroon, Emerg. Infect. Dis. 11, 1935–1938 (2005)
J.C. Quixabeira-Santos, M.L.G. Medaglia, C.A. Pescador, C.R. Damaso, Emerg. Infect. Dis. 17, 726–729 (2011)
A.T. Silva-Fernandes, C.E. Travassos, J.M. Ferreira, J.S. Abrahão, E.S. Rocha, F. Viana-Ferreira, J.R. dos Santos, C.A. Bonjardim, P.C. Ferreira, E.G. Kroon, J. Clin. Virol. 44(4), 308–313 (2009)
G.S. Trindade, G.L. Emerson, D.S. Carroll, E.G. Kroon, I.K. Damon, Emerg. Infect. Dis. 13(7), 965–972 (2007)
F. Fenner, D.A. Henderson, T. Arita, Jezek, I.D. Ladnyi, Geneva, WHO. ISBN 92-4-156110-6 (1988)
G. McFadden, Nat. Rev. Microbiol. 3, 201–213 (2005)
H.W. Chang, J.C. Watson, B.L. Jacobs, Proc. Natl. Acad. Sci. 89, 4825–4829 (1992)
C.K. Ho, S. Shuman, Virology 217, 272–284 (1996)
C. Rivas, J. Gil, Z. Melkova, M. Esteban, M. Diaz-Guerra, Virology 243, 406–414 (1998)
S. Guerra, A. Ca′ceres, K.P. Knobeloch, I. Horak, M. Esteban, PLoS. Pathog. 4, 1–16 (2008)
E. Beattie, J. Tartaglia, E. Paoletti, Virology 183, 419–422 (1991)
E. Beattie, E. Paoletti, J. Tartaglia, Virology 210, 254–263 (1995)
M.V. Davies, O. Elroy-Stein, R. Jagus, B. Moss, R.J. Kaufman, J. Virol. 66, 1943–1950 (1992)
C. Gubser, S. Hue, P. Kellam, G.L. Smith, J. Gen. Virol. 85, 105–117 (2004)
J.L. Najera, C.E. Gomez, E. Domingo-Gil, M.M. Gherardi, M.J. Esteban, Virology 80, 6033–6047 (2006)
X. Meng, C. Jiang, J. Arsenio, K. Dick, J. Cao, Y. Xiang, J. Virol. 83, 10627–10636 (2009)
L.G. Payne, J. Gen. Virol. 50, 89–100 (1980)
S.N. Isaacs, E.J. Wolffe, L.G. Payne, B. Moss, J. Virol. 66, 7217–7224 (1992)
E. Herrera, M.D.M. Lorenzo, R. Blasco, S.N. Isaacs, J. Virol. 72(1), 294–302 (1998)
E.J. Wolffe, S.N. Isaacs, B. Moss, J. Virol. 67, 4732–4741 (1993)
I.P. Singh, S.B. Singh, J. Res. Ludhiana 4, 440–448 (1967)
S.F. Altschul, T.L. Madden, A.A. Schaffer, J. Zhang, Z. Zhang, W. Miller, D.J. Lipman, Nucleic Acids Res. 25, 3389–3402 (1997)
K. Tamura, D. Peterson, N. Peterson, G. Stechter, M. Nei, S. Kumar, Mol. Biol. Evol. (2011). doi:10.1093/molbev/msr121
M.J.R. Gale, S.L. Tan, M. Wambach, M.G. Katze, Mol. Cell. Biol. 16(8), 4172–4181 (1996)
M. Kawagishi-Kobayashi, J.B. Silverman, T.L. Ung, T.E. Dever, Mol. Cell. Biol. 17, 4146–4158 (1997)
A.C. Dar, F. Sicheri, Mol. Cell 10, 295–305 (2002)
S. Bakes, K.M. Sperling, J. Zwilling, G. Gasteiger, H. Ludwig, E. Kremmer, A. Schwantes, C. Staib, G. Sutter, J. Gen. Virol. 91, 470–482 (2010)
G.S. Trindade, Z.I.P. Lobato, B.P. Drumond, J.A. Leite, R.C. Trigueiro, M.I.M.C. Guedes, F.G. da Fonseca, J.R. dos Santos, C.A. Bonjardim, P.C.P. Ferreira, E.G. Kroon, Am. J. Trop. Med. Hyg. 75, 486–490 (2006)
G. Li, N. Chen, Z. Feng, R.M.L. Buller, J. Osborne, T. Harms, I. Damon, C. Upton, D.J. Esteban, Virol. J. 3, 88–97 (2006)
F. Fenner, D.A. Henderson, I. Arita, Z. Jezek, I.D. Ladnyi, World Health Organisation, Geneva, 1988
Q. Li, C. Upton, B. Hazes, D.H. Evans, J. Virol. 85, 1349–1360 (2011)
R.K. Singh, V. Balamurugan, M. Hosamani, B.M. Chandra Naik, G. Krishnappa, M.P. Yadav, Acta Virol. 51, 47–50 (2007)
M.H.V. Van Regenmortel, C.M. Fauquet, D.H.L. Bishop, Virus Taxonomy: The Classification and Nomenclature of Viruses. The seventh report of the international committee on taxonomy of viruses. (Academic Press, San Diego, 2000). p. 1167
Acknowledgments
The authors are thankful to the Indian Council of Agricultural Research for financial assistance and providing the necessary facilities to carry out the study.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Bera, B.C., Shanmugasundaram, K., Barua, S. et al. Sequence and phylogenetic analysis of host-range (E3L, K3L, and C7L) and structural protein (B5R) genes of buffalopox virus isolates from buffalo, cattle, and human in India. Virus Genes 45, 488–498 (2012). https://doi.org/10.1007/s11262-012-0788-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-012-0788-8