
Abstract
Rabbit hemorrhagic disease is an acute fatal highly contagious viral infectious disease that causes high losses among rabbitries. The disease was first reported in China in 1984 and later on in Saudi Arabia in 1996. The aim of this study was to investigate the emergence and pathogenicity of new rabbit hemorrhagic disease virus (RHDV) strains in Saudi Arabia. The pathogenicity was confirmed by inoculation in susceptible rabbits. Three RHDV strains were detected by reverse transcriptase polymerase chain reaction (RT-PCR) using primers targeting VP60 capsid protein gene in infected rabbitries during 2012 and 2013. These strains clustered into two genetically distinct genogroups related to year of isolation (G2 and G3). All new Saudi Arabia viruses clustered with the European strains, while the old strains clustered with strains from China and America. Based on amino acids and nucleotide sequences, the Saudi Arabia strains (RHD/1/SA/2012, RHD/2/SA/2012, and RHD/3/SA /2013) had high identity with Mexico89, Ca11-ITA, and 00–13,FRA virus; on the other hand, there was a relatively high identity with Bahrain strain. The evolutionary relationship of Saudi RHDVs strains revealed significant nucleotides and amino acid substitutions in hypervariable region E, suggesting the emergence of new RHDVs circulating in Saudi Arabia rabbitries. These antigenic changes represented by the antigenic index might be a potential cause of vaccination failure and raises the need to review the vaccination strategies against RHD.
Similar content being viewed by others


Introduction
Rabbit hemorrhagic disease (RHD) is a fatal and contagious viral disease affecting both domesticated and wild rabbits and causes high economic losses and mortalities ranging from 45 to 100% (Marchandeau et al. 1998; OIE 2012; Fuller et al. 1993). The disease was first reported in China in 1980s (Liu et al. 1984). Within 1 year, the disease spread and killed 140 million domestic rabbits in China (Xu 1991). The disease spreads to Europe and was first detected in Italy then in several European countries and became a pandemic with several lethal outbreaks occurring in many countries (Berninger and House 1995; Cancellotti and Renzi 1991). The disease was first reported in Saudi Arabia and killed 100% of rabbits in rabbitries during November 1996 (Abu Elzein and Al Afaleq 1999). Five years later (May 2001), an outbreak was recorded in Bahrain Kingdom at a private farm that lasted for 2 weeks and resulted in high mortalities (Forrester et al. 2006). The disease is characterized by fever, dullness, and prostration followed by death after short incubation period ranging from 1 to 3 days.
Typical lesions are liver necrosis, enlarged spleen, and hemothorax (Lavassa and Capucci 2008). The etiological agent, rabbit hemorrhagic disease virus (RHDV), belongs to genus Lagovirus of family Caliciviridae (Koopmans et al. 2005). The virus genome is a positive sense ssRNA of 7.2 kb. The virus is not cultivable in vitro; therefore, genomic detection of the virus, antibody detection, and experimental infection of serum-negative rabbits are needed for diagnosis and characterization (OIE 2012). One of the major proteins encoded by RHDV genome is the VP60 protein which contains the hypervariable region E: a type-specific antigenic epitope (Capucci et al. 1998). Phylogenetic studies have been published to evaluate the genetic variations between RHDVs based on partial and complete sequence of VP60 capsid protein (Forrester et al. 2006, 2009; Le Gall-Reculé et al. 2003; McIntosh et al. 2007; Wang et al. 2012). The aims of this study were to identify and analyze the evolutionary changes of the newly emerged Saudi Arabia RHDVs detected during 2012–2013.
Materials and methods
Samples
Between 2012 and 2013, 96 rabbits (3–4 months old) from three geographically distinct rabbitries in Al-Hassa region of Saudi Arabia were presented to The Veterinary Teaching Hospital (VTH), College of Veterinary Medicine, King Faisal University. The rabbits had signs of infection with RHD. The suspected diseases’ outbreaks occurred in April 2012 (two rabbitries) and in May 2013 (one rabbitry). Dead and sacrificed diseased rabbits were subjected for clinical and postmortem examination. Five liver samples were collected from each herd from sacrificed and/or dead rabbits suspected to be infected with RHD. Samples from each herd were pooled and processed as described by OIE manual (2012). The homogenates were stored at −80 °C until used.
Experimental inoculation for pathogenicity
Twelve New Zealand white 4-month-old susceptible rabbits were allotted randomly into four equal groups. Rabbits of groups 1, 2, and 3 were inoculated intramuscularly with 1 ml of 10% w/v filtered RHDV-infected liver homogenate of RHD/1/SA/2012, RHD/2/SA/2012, and RHD/3/SA/2013, respectively, while group 4 was kept as a negative control. All groups were kept separately for 3 weeks under the same management system. Clinical signs and mortalities were recorded. Dead rabbits were necropsied and gross lesions recorded, while surviving rabbits in each group were sacrificed 21 days post-inoculation. Three liver samples from each group were pooled, processed, and prepared for virus detection.
RNA extraction
Total RNA was extracted with QIAamp Viral RNA Mini Kit (QIAGEN, USA) from liver homogenates according to the manufacturer’s recommendation. Briefly, 140 μl of the liver homogenates was lysed by adding 560 μl of AVL buffer containing carrier RNA, after complete lysis 500 μl of absolute ethanol was added and mixed for 15 s by pulse vortexing. Aliquots of 630 μl were transferred sequentially to a QIAamp spin column followed by centrifugation at 8000 rpm for 1 min after each addition. The binding RNAs were washed by the addition of 500-μl AW1 buffer and centrifuged at 8000 rpm for 1 min followed by the addition of 500 μl AW2 buffer then centrifuged at 14,000 rpm for 3 min. The RNAs were eluted in 60 μl of buffer AVE and stored at −85 °C until used.
Amplification and sequencing of VP60
One-step RT-PCR kit (QIAGEN, USA) was used to amplify VP60 of the RHDV genome. The specific oligonucleotide primers, RHD-F 5′-ATGGAGGGCA AAGCCCGCACAGCG-3′ and RHD-R 5′-AATTCAGACATAAGAAAAGCCA TTG-3′ were used to amplify the 1740 bp coding region for the VP60 capsid protein. The RT-PCR reaction mixture consisted of 5 μl of the total RNA, 10 μl 5× Qiagen one-step RT-PCR buffer, 10 μl Q buffer, 2 μl of dNTPs mix, 1 μl (50 pmol) of each primers, 2 μl of the enzyme mix (containing RT and PCR reaction enzymes), and 19 μl of RNase free water. The RT-PCR reaction was performed at 50 °C for 30 min, then 95 °C for 15 min, followed by 40 cycles at 95 °C for 1 min, 55 °C for 30 s and 72 °C for 1 min, and a final extension step at 72 °C for 10 min. The PCR products were analyzed on 1.25% agarose gel containing 0.5 μg/ml ethidium bromide and visualized under UV light. The PCR products were excised from the agarose gel then purified using Montàge DNA gel extraction kit (Millipore, USA) and sequenced in an automated ABI 3730 DNA sequencer (Applied Biosystems, USA).
Phylogenetic analysis
The obtained sequence data were analyzed using online BLAST server and compared with RHDV sequences (partial or complete) available in GenBank. Multiple sequence alignment was carried out using Lasergene sequence analysis software (DNASTAR, USA). A phylogenetic tree was constructed by neighbor-joining method using PAUP*, version 4.0 (Swofford 2000). Bootstrap values were calculated for 1000 replicates of the alignment.
GenBank accession number
The obtained VP60 gene sequences of the detected RHDVs were submitted to Genbank database with the accession numbers ((KJ949619) RHD/1/SA/2012, (KJ949620) RHD/2/S A/2012, and (KJ949621) RHD/3/SA/2013).
Results
Clinical examination
Diseased rabbits from the three rabbitries suffered from bloody nasal discharge, weight loss, and lethargy signs continue for 5–7 days with high morbidity ranged from 60 to 70% followed by high mortality rate ranging from 80 to 100% of infected rabbits. In addition, the postmortem examination of dead or sacrificed rabbits revealed pneumonia and hemothorax. These signs were suggestive for RHDV infection. The virus VP60 capsid protein gene was successfully detected using RT-PCR in all tested herds.
Experimental inoculation
Inoculated rabbits developed high fever (39.7 °C) within 24 h post-inoculation continued for 3 days. Signs in groups 1, 2, and 3 were characterized. Generally, the signs in all inoculated groups were similar, progressed to depression and bloody nasal discharges. Mortalities reached 66.6 to 100% in different groups. All rabbits in groups 1 and 3 died within 4–5 days PI. In group 2, only one rabbit survived without showing clinical signs and sacrificed 21 days PI. Post-mortem examination revealed enlarged liver, splenomegaly, hemorrhage in the trachea, hemothorax, and pneumonia with different severity in all dead rabbits in different groups. No changes were observed at necropsy in the asymptomatic surviving rabbit and uninoculated controls. Liver homogenates prepared from the all dead rabbits were positive for VP60 capsid protein detection, while the survived asymptomatic rabbit (group 2) and all samples from the group 4 (Negative control group) were negative for viral capsid protein VP60 using RT-PCR.
VP60 detection and genomic sequence analysis
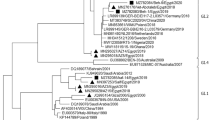
The obtained PCR products (1740 bp) were excised, purified, and sequenced. The VP60 gene sequence data were submitted to Genbank and aligned with 52 RHDVs that represent different geographical areas and multiple outbreaks (Fig. 1). The current Saudi RHDVs were closely related to each other with identity percentage ranging from 99.7 to 99.9%. Meanwhile, these viruses were also closely related to RHDVs isolated from Italy, Mexico, and France (Ca11-ITA, Mexico89-MEX and 00-013-FRA) with identity ranged from 93.6 to 96%. These newly emerging RHDVs diverged widely from the old Saudi strains isolated during 1996’s outbreak; the divergence was 12% approximately (Table 1). In addition, the new Saudi strains were found to be genetically different from the RHDV isolated from Bahrain Kingdom (DQ189077) during 2001 outbreak with an identity percentage ranging from 90.7 to 90.8%.

Phylogenetic analysis of partial VP60 capsid protein sequence for 54 RHDVs strains representing different geographical areas. Branch lengths correlated to genetic distance. RCV, ITA, and Ashington, UK, were used as an out-group to root of the tree. Bootstrap values for 1000 replicate calculated using neighbor-joining algorithm, for clarity only values, more than 50 are shown
The deduced amino acids sequences of the newly emerging Saudi strains showed a major identity (100%) with isolates from Italy, France, and Mexico. The deduced amino acid sequences analysis proved heterogeneity between the new and old Saudi strains with divergence percentage ∼13%. Although these newly emerging Saudi strains had relative heterogeneity with RHDV detected in Bahrain Kingdom (DQ189077) at the nucleotide level (Table 1), the deduced amino acid sequences revealed a minor heterogeneity with identity percentage ranging from 98.1 to 98.5%. Antigenic index illustrated different peak shape among new, old Saudi, and other RHDVs.
Phylogenetic relationship of Saudi RHDVs
For simplicity, the aligned 54 RHDVs identified into four distinct geno-groups (Fig. 1) on bases of bootstrapping and clustering. Genogroup 1 contains the genetically divergent viruses (RCV, ITA, and Ashington,UK) which were used as the out-group in the phylogenetic tree. The old Saudi RHDVs isolated during 1996 outbreak were categorized with viruses from different geographical areas representing different world continents in genogroup 2. These viruses are the direct descendants from Hartmannsdorf, DEU. The newly emerged Saudi RHDVs isolated during 2012–2013 clustered with European viruses that were associated with several epidemics in Europe during the last decade as well as the Mexican strain Mexico89 in genogroup 3. In addition, the Bahrain strain which caused a fatal outbreak in Bahrain Kingdom during 2001 was grouped with the newly emerged Saudi RHDVs in genogroup 3. Viruses from Spain and Portugal exclusively clustered together in a distinct lineage with some viruses from Egypt, France, and Czech Republic in genogroup 4 with WX/China/84,CHN that considered the first RHDV detected worldwide. Our phylogenetic analysis showed certain significance of geographical location and/or date of isolation among viruses in each genogroup.
Evolutionary changes in new RHDVs in reliability of old Saudi RHDVs
Deduced amino acid sequence of VP60 of our new Saudi RHDVs was aligned with the old Saudi viruses and representative reference viruses from different geographical areas. Phylogenic analysis of the deduced amino acid sequence showed that the new Saudi viruses could be placed in a separate clade in a distinct cluster (Fig. 2). We compared the deduced amino acids sequence of VP60 within residue 340 to 440 including the hypervariable region E in the capsid protein. Thirteen amino acid substitutions were found between recent Saudi viruses and old (classical) strains, which almost has the same amino acid sequence of strain (Kal2005) detected in Egypt. The new Saudi strains shared the same amino acid substitutions with (Ca11,ITA; Mexico89, MEX, and 00–13,FRA). A single amino acid substitution was found between our recent viruses and the virus isolated from Bahrain Kingdom (Table 2). The recent Saudi strains showed identical antigenic peaks with Bahrain strain and were quite identical to Mexico89, MEX. Whereas, the classical Saudi strains showed certain differences in the antigenic peaks with the recent strains according to antigenic index, Jamesone-wolf protean analysis, and DNASTAR software package.

Phylogenetic relationship of 20 RHDVs based on deduced amino acid sequences of VP60 capsid protein. RCV, ITA (black circle) used as out-group to root the tree. (black triangle) Old Saudi strains, and (black square) New Saudi strains. Bootstrapping values are placed on main branches for clarity
Discussion
We present a comparative molecular analysis of the newly emerged RHDVs in Saudi Arabia during 2012–2013 in comparison with the old RHDVs from the first recognized outbreak in 1996 based on VP60 capsid protein sequences. In these recent field outbreaks in Saudi Arabia rabbitries, the clinical and gross lesions in combination with disease reproduction in a small-scale experiment using susceptible rabbits proved that the detected RHDVs were virulent and produced high losses. The reported clinical signs in our study showed some variations from the previously reported 2001 outbreak in Bahrain Kingdom (Forrester et al. 2006); the lesions were clearer and prominent that may be due to breed variation, age, susceptibility, and pathogenicity of the virus also from natural to experimental infection (Lavassa and Capucci 2008). Our results were supported by the successful amplification of VP60 capsid protein using RT-PCR followed by genomic sequencing. Based on genomic sequence of VP60 capsid protein, we conclude that there is a significant genomic difference between the Saudi RHDVs over a period of 16 years. The divergence percentage between the recent Saudi RHDVs (2012–2013) and old Saudi RHDVs (1996) is approximately 12%. The maximum divergence between old and recent viruses detected in France ranged from 7.6 to 8.5% over a period of 11 years as identified by Le Gall et al. 1998; Le Gall-Reculé et al. 2003. The much lower nucleotide diversity (less than 4%) between the recent Saudi RHDVs and viruses detected in France and Mexico suggests that these viruses are closely related to each other. Consequently, an epidemiological connection exists between Saudi Arabia, France, and Mexico resulting in spread from one country to another through animal as well as animal byproducts trading or that the viruses spreading to these countries came from a common source. For clarity, we have divided RHDVs into four genogroups based on the nucleotide sequence of VP60 capsid protein and bootstrapping results according to previous reports (Abrantes et al. 2012; Alda et al. 2010; Tamura et al. 2013). The avirulent Italian isolate (RCV) and Ashington virus, UK, were placed in a separate group (G1) as they are antigenically related but genetically distinct from each other and from all other RHDVs (Forrester et al. 2006; Kerr et al. 2009; Moss et al. 2002). The Saudi RHDVs grouped into two different genogroups (G2 and G3) according to the year of isolation, the same pattern observed among French viruses as indicated by (Le Gall-Reculé et al. 2003). The presented phylogenic analysis has indicated that the recent Saudi RHDVs clustered in the same clade with other European viruses isolated during the last decade. In addition, Mexico89 strain isolated from Mexico since more than two decades fall in the same cluster with a minimum divergence with our isolates and 100% nucleotide identity with the 00–13,FRA possibly due to the virus reemerging as indicated by (Wang et al. 2012). Bahrain RHDV isolated during 2001 showed more amino acid homology (approximately ∼99%) with the new Saudi viruses, possibly due to the geographical proximity between Kingdom of Bahrain and Saudi Arabia. A significant correlation between individual viruses isolated from a certain geographical area was obvious in our genogroups that was probably due to the efficient distribution of the viruses by human and commercial activities as indicated by (Moss et al. 2002). The old Saudi Arabia RHDVs isolated during 1996 outbreak composed a distinct cluster with other RHDVs from China, Japan, and USA. We documented a number of amino acid substitutions among the recent and classical Saudi viruses compared to reference RHDVs (Table 2). The recent Saudi viruses showed identical amino acid sequence to Ca11,ITA; Mexico, MEX, and 00–13,FRA viruses. Interestingly, using the avirulent Italian strain RCV as out group to root the tree based on amino acid sequences, the recent viruses were included in a separate lineage containing the original Chinese strain WX/China/84,CHN isolated from the first recorded outbreak (Wang et al. 2012). Although, they were in two separate clusters at the nucleotide level, this presumably due to silent mutations in the coding region of the genome. The amino acid substitution among various strains affect the antigenic beaks of RHDVs that may lead to vaccination failure in certain circumstances as previously reported by (Wang et al. 2012). RHDVs previously distinguished as original RHDV and (RHDVa) variant RHDVs (McIntosh et al. 2007). Based on monoclonal antibodies ELISA combined with amino acid sequence analysis, RHDVs might be grouped as original and variant viruses. Interestingly, our phylogenic tree and analysis of other published data revealed that the classification of some strains as original or variant viruses could be changed if aligned with different virus sequences (i.e., the old Saudi viruses placed by (McIntosh et al. 2007) as original viruses, fall in same cluster with some of variant viruses). In addition, lack of time-related distribution between original and variant viruses that may raise the hypothesis that the old viruses are not replaced by the new viruses and viruses could be re-emerged as also concluded by (Forrester et al. 2006; Wang et al. 2012; Schwensow et al. 2014). In conclusion, comprehensive analysis of the new RHDVs detected during 2012 and 2013 outbreaks was described. The new viruses formed a distinctive group from the classical Saudi viruses. On the other hand, these recent viruses were clustered with European viruses that caused many epidemics last decade. It is notable that antigenic variant Saudi RHDVs have been emerged exhibiting a significant antigenic variations from the classical Saudi strains on bases of nucleotide and amino acid sequence. Interestingly, the recent Saudi viruses shared a complete amino acid sequence identity with the Ca11,ITA; Mexican89, MEX, and 00–13,FRA strains as well as a high identity with WX/China/84,CHN; this suggested that these viruses have maintained in the population circulated harmlessly and might be re-emerged as virulent strains. Finally, we suggest finding more applicable and clear basics to delineate the genetic and antigenic properties of these viruses to distinguish variant from classical strains.
References
Abrantes, J., van der Loo W., Le Pendu, J. and Esteves P., 2012. Rabbit haemorrhagic disease (RHD) and rabbit haemorrhagic disease virus (RHDV): a review. Veterinary Research, 43, 12.
Abu Elzein, E.M. and Al Afaleq, A.I., 1999. Rabbit haemorrhagic disease in Saudi Arabia. Veterinary Records, 144 (17), 480–481.
Alda, F.; Gaitero, T.; Saurez, M.; Rocha, G. and Doadrio, I., 2010. Evolutionary history and molecular epidemiology of rabbit haemorrhagic disease virus in Iberian Peninsula and Western Europe. BMC Evolutionary Biology 10: 347.
Berninger, M.L. and House, C., 1995. Serologic comparison of four isolates of rabbit hemorrhagic disease virus. Veterinary Microbiology, 47 (1–2),157–165.
Cancellotti, F.M. and Renzi, M., 1991. Epidemiology and current situation of viral haemorrhagic disease of rabbits and the European brown hare syndrome in Italy. Revue scientifique et technique, 10,409–422.
Capucci, L., Fallacara, F., Grazioli, S., Lavazza, A., Pacciarimi, M.L. and Procchi, E. 1998., A further step in the evolution of rabbit hemorrhagic disease virus: the appearance of the first consistent antigenic variant. Virus Research, 58, 115-126.
Forrester, N.L., Abubakr, M.I., Abu Elzein, E.M., Al-Afaleq, A.I., Housawi, F.M., Moss, S.R., Turner, S.L. and Gould, E.A., 2006. Phylogenetic analysis of rabbit haemorrhagic disease virus strains from the Arabian Peninsula: did RHDV emerge simultaneously in Europe and Asia? Virology, 344, 277–282.
Forrester, N.L. Boag, B., Buckley, A., Moureau, G. and Gould, E.A., 2009. Co-circulation of widely disparate strains of rabbit haemorrhagic disease virus could explain localised epidemicity in the United Kingdom. Virology, 393, 42-48.
Fuller, H.E., Chasey, D., Lucas, M.H. and Gibbens, J.C., 1993. Rabbit haemorrhagic disease in the United Kingdom. Veterinary Record, 133, 611–613.
Kerr, P.J., Kitchen, A. and Holmes, E.C., 2009. Origin and phylodynamics of rabbit hemorrhagic disease virus. Journal of Virology, 83, 12129–12138.
Koopmans, M.K., Green, K.Y., Ando, T., Clarke, I.N., and Estes, M.K., 2005. Family Caliciviridae. In: Virus Taxonomy: Eighth Report of the International Committee on Taxonomy of Viruses, eds C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger & L. A., Ball Academic Press Inc., London.
Lavassa, A. and Capucci, L., 2008. How many caliciviruses are there in rabbits. A review on RHDV and correlated viruses. In: LagomorphBiology: Evolution, Ecology and Conservation. Springer-Verlag Berlin Heidelberg, 263–278.
Le Gall, G., Arnauld, C., Boilletot, E., Morisse, J.P. and Rasschaert, D., 1998. Molecular epidemiology of rabbit haemorrhagic disease virus outbreaks in France during 1988 to 1995. Journal of General Virology, 79, 11–16.
Le Gall-Reculé G., Zwingelstein, F., Laurent, S., de Boisséson, C., Portejoie, Y. and Rasschaert, D., 2003. Phylogenetic analysis of rabbit haemorrhagic disease virus in France between 1993 and 2000, and the characterisation of RHDV antigenic variants. Archives of Virology, 148 (1), 65-81.
Liu, S.J., Xue, H.P., Pu, B.Q. and Qian, N.H. 1984. A new viral disease in rabbits. Animal Husbandry and Veterinary Medicine, 16, 253–255.
Marchandeau, S., Chantal, J., Portejoie, Y., Barraud, S. and Chaval, Y., 1998. Impact of viral haemorrhagic disease on a wild population of European rabbits in France. Journal of Wild Diseases 34, 429–435.
McIntosh, M.T., Behan, S.C., Mohamed, F.M., Lu, Z., Moran, K.E., Burrage, T.G., Neilan, J.G., Ward, G.B., Botti, G., Capucci, L. and Metwally, S.A., 2007. A pandemic strain of calicivirus threatens rabbit industries in the Americas. Virology Journal, 2, 94:96.
Moss, S.R., Turner, S.L., Trout, R.C., White, P.J., Hudson, P.J., Desai, A., and Armesto M., 2002. Molecular epidemiology of Rabbit haemorrhagic disease virus. Journal of General Virology, 83, 2461–2467.
Schwensow, N. I., Cooke, B., Kovaliski, J., Sinclair, R., Peacock, D., Fickel, J. and Summer S., 2014. Rabbit haemorrhagic disease: virus persistence and adaptation in Australia. Evolutionary Applications, 7, 1056–1067.
Swofford, D.L., 2000. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Version 4. Sinauer Associates, Sunderland, Massachusetts.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S., 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729.
Wang, X., Hao, H., Qiu, L., Dang, R., Du, E., Zhang, S. and Yang, Z., 2012. Phylogenetic analysis of rabbit hemorrhagic disease virus in China and the antigenic variation of new strains. Archives of Virology, 157(8), 1523-1530.
World Organization for Animal Health. Terrestrial manual (OIE), 2012 Chapter2.6.2. Rabbit haemorrhagic disease. http://www.oie.int/fileadmin/Home/eng/Health_standards/tahm/2.06.02_RHD.pdf.
Xu, W.Y., 1991.Viral haemorrhagic disease of rabbits in the People’s Republic of China: epidemiology and virus characterization. Revue scientifique et technique, 10, 393–408.
Acknowledgments
We thank Deanship of Scientific Research, King Faisal University for supporting this research article [Project No. 130265].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Ismail, M.M., Mohamed, M.H.A., El-Sabagh, I.M. et al. Emergence of new virulent rabbit hemorrhagic disease virus strains in Saudi Arabia. Trop Anim Health Prod 49, 295–301 (2017). https://doi.org/10.1007/s11250-016-1192-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11250-016-1192-5