
Abstract
Porcine epidemic diarrhea virus (PEDV) belongs to the Coronaviridae family and causes acute enteritis in pigs. A fragment of the large spike glycoprotein, termed the S1D epitope (aa 636–789), alone and fused with cholera toxin B subunit, were independently cloned into plant expression vectors, yielding plasmids pMYV717 and pMYV719, respectively. Plant expression vectors were transformed into Agrobacterium tumefaciens and subsequently infiltrated into Nicotiana benthamiana leaves. The highest expression level of S1D was found at 2 days post infiltration (dpi), reached 0.04 % of total soluble protein, and rapidly decreased thereafter. The expression and assembly of CTB–S1D fusion protein were confirmed by Western blot and GM1-ELISA. The highest expression level of CTB–S1D fusion protein was 0.07 % of TSP at 4 dpi, with a rapid decrease thereafter. In the presence of p19 protein from tomato bushy stunt virus, the S1D and CTB–S1D protein levels peaked at 6 dpi and were fourfold to sevenfold higher than in the absence of p19, respectively. After oral administration of transiently expressed CTB–S1D fusion protein, or with bacterial cholera toxin or rice callus expressing mutant cholera toxin 61F, mice exhibited significantly greater serum IgG and sIgA levels against bacterial CTB and S1D antigen, peaking at week 6. Transiently expressed CTB–S1D fusion protein will be administered orally to pigs to assess the immune response against PEDV.
Similar content being viewed by others

Introduction
Vaccines have been used to prevent a wide range of infectious and non-infectious diseases (Poland et al. 2002). Vaccine antigens are composed of antigenic proteins and are devoid of pathogenic genes, so cannot establish infection (Strugnell et al. 2011). Vaccine antigens can be produced using a variety of host expression systems, including genetically engineered bacterial, yeast, mammalian and plant cells. Plants are attractive expression vehicles for commercial production of vaccine antigens. Plant-based vaccines hold great promise as they are cost-effective, easy-to-administer, easy-to-store, fail-safe and acceptable, particularly in developing countries (Lal et al. 2007; Sala et al. 2003). Novel approaches to improve the yield and biosafety of plant-based vaccines have been proposed (Hernández et al. 2014). Agroinfiltration is a simple, efficient and robust method for transient protein expression (Leuzinger et al. 2013).
Porcine epidemic diarrhea virus (PEDV), classified as a member of group I of the genus Alphacoronavirus, the family Coronaviridae, and the order Nidovirales, causes porcine epidemic diarrhea (PED) in pigs of all ages, especially in newborn pigs (Pensaert and de Bouck 1978). PEDV disrupts villus enterocytes and causes villi atrophy within the jejunum and ileum, leading to a mortality rate of up to 95 % in infected suckling pigs (Ducatelle et al. 1981). PED was first detected in Belgium (Pensaert and de Bouck 1978) and the UK (Chasey and Cartwright 1978), and was designated CV777. Outbreaks of this disease, which have been reported in many pig farming countries, have led to severe economic losses in Canada (Turgeon et al. 1980), Japan (Takahashi et al. 1983), China (Li et al. 2012), Thailand (Puranaveja et al. 2009), Korea (Lee and Lee 2014), the United States (Stevenson et al. 2013) and, more recently, in Austria (Steinrigl et al. 2015).
The genome of PEDV consists of a single molecule of linear positive-sense single-stranded RNA. The complete sequence of the genome of strain CV777 was found to be 28,033 nucleotides in length, excluding the poly A-tail (Kocherhans et al. 2001). The large spike glycoprotein peplomer (S), one of four structural proteins, stimulates production of neutralizing antibodies in the host. The predicted S polypeptide is 1383 amino acids long, contains 29 potential N-linked glycosylation sites and shows structural features similar to those of the coronavirus spike protein (Duarte and Laude 1994). The PEDV S protein can be divided into the S1 domain (aa 1–789) and S2 domain (aa 790–1383) based on the presence of the conserved nonamer and GxCx motifs at the proteolytic cleavage site of the S protein of other members of coronavirus group II (Follis et al. 2006). Within the S1 domain, two important epitope domains were determined. The first domain is the PEDV-neutralizing epitope (CO-26 K equivalent, COE gene), which is 139 aa long and spans the region aa 499–638 (Chang et al. 2002). Sun et al. identified the second epitope domain, designated S1D (aa 636–789), and reported that it induces neutralizing antibodies (Sun et al. 2008). In this study, the S1D gene alone, and that fused with the cholera toxin B subunit (CTB) gene of Vibrio cholerae, were cloned into plant expression vectors. These vectors were transiently expressed in Nicotiana benthamiana (N. benthamiana) using agroinfiltration. The proteins were orally administered to mice to induce specific immune responses.
Materials and methods
Construction of S1D and a CTB–S1D fusion gene for transient expression in N. benthamiana
To construct an S1D gene for transient expression in N. benthamiana, plasmid pMYV98 containing the spike protein gene of PEDV was used as a template for PCR. A pair of primers (forward primer 5′-GGATCC GAC GTT TCT TTT ATG AC-3′ and reverse primer 5′-GGTACC AAT ACT CAT ACT AAA G-3′) was designed to amplify the S1D gene. A BamHI restriction site at the 5′-end and KpnI at the 3′-end of the sequence was included to facilitate cloning. The S1D gene was amplified using Ex Taq (Takara Bio, Shiga, Japan) with the following PCR conditions: one cycle at 94 °C for 5 min; 30 cycles at 94 °C for 30 s, 58 °C for 30 s and 72 °C for 30 s, followed by one cycle at 72 °C for 5 min. The products were cloned into the pGEM®T-Easy vector (Promega, Madison, WI, USA), creating plasmid pMYV712. Gene identity was confirmed via sequencing using the universal primers T7 and SP6. The BamHI and KpnI digested fragment containing the S1D gene from pMYV712 was introduced into the same sites of digested plant-expression vector pMYV497 under the regulation of the duplicated Cauliflower mosaic viral 35S promoter (dp35S), CTB signal peptide, ER retention signal (SEKDEL) (Munro and Pelham 1987) and Nos-T, yielding pMYV717. pMYV508 harboring the p19 protein of tomato bushy stunt virus (TBSV), which prevents post-transcriptional gene silencing (PTGS) in infiltrated tissues, was used for co-expression (Voinnet et al. 2003).
To construct the CTB–S1D fusion gene, the BamHI and KpnI-digested fragment containing the S1D gene from plasmid pMYV712 was inserted into the same sites of plasmid pMYV498 to generate plasmid pMYV719. This plasmid contains dp35S, CTB adjuvant fused with S1D at the N-terminus, an ER retention signal (SEKDEL) and Nos-T. Plasmids pMYV717 and pMYV719 were transformed into Agrobacterium tumefaciens strain LBA4404 together with the helper plasmid pRK2013 using the tri-parental mating method (Horsch et al. 1985).
Construction of the S1D gene for expression in E. coli and production of mouse anti-S1D antibody
The plasmid pMYV98 containing the spike protein gene from PEDV was used as a template for PCR. A pair of primers (forward primer 5′-GGATCC GAC GTT TCT TTT ATG AC-3′ and reverse primer 5′-GGTACCTTAAAT ACT CAT ACT AAA G-3′) was designed to amplify a PCR fragment containing the S1D gene and a stop codon (TAA) upstream of the KpnI enzyme site. A BamHI restriction site at the 5′-end and KpnI at the 3′-end of the sequence was included to facilitate cloning. The S1D gene for expression in E. coli was amplified using Ex Taq (Takara Bio) with the PCR conditions described above and cloned into the pGEM®T-Easy vector (Promega), creating plasmid pMYV711. Gene identity was confirmed via sequencing using the universal primers T7 and SP6. The BamHI and KpnI-digested fragment containing the S1D gene from pMYV711 was introduced into the same sites of the E. coli expression vector pQE-30 (Qiagen, Hilden, Germany), yielding pMYV714. The plasmid was confirmed by restriction enzyme mapping. Plasmid pMYV714 was transformed into E. coli expression host strain SG13009 (Qiagen) for production of recombination protein.
Purification of the recombinant S1D protein synthesized in E. coli was performed under denaturating conditions in 8 M urea (Kim et al. 2009). Briefly, a bacterial colony harboring the S1D gene was inoculated into 5 mL of Luria Bertani (LB) medium containing ampicillin (100 mg/l) and kanamycin (5 mg/L), and incubated overnight at 37 °C. The culture was transferred to 200 mL of LB medium and incubation continued at 37 °C for 2 h to an OD600 of 0.6–0.8. Expression of the recombinant proteins was induced by adding iso-propyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 10 mM, followed by incubation for a further 6 h at 37 °C. The cells were harvested by centrifugation and lysed in 10 mL of ‘buffer Z’ (8 M urea, 100 mM NaCl, 20 mM HEPES, pH 8.0) by sonication on ice (20 min; 20 s runs with 15 s breaks between each run). After centrifugation at 10,000 rpm for 10 min at 4 °C using a JA-14 rotor (Beckman Coulter, Pasadena, CA, USA) to remove cell debris, imidazole was added to the bacterial lysate supernatant to a final concentration of 10 mM and the sample was loaded onto a 2 mL nickel column (Ni–NTA; Invitrogen, Carlsbad, CA, USA). The histidine-affinity column was washed with 15 mL of buffer Z plus imidazole (10 mM) to remove weakly bound proteins of E. coli origin. The His-tagged recombinant proteins were eluted with buffer Z plus 250 mM imidazole. The purified recombinant proteins were quantified by Bradford protein assays (Bio-Rad, Hercules, CA, USA) and dialyzed in phosphate-buffered saline (PBS) containing 8.0 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4.2 H2O and 0.24 g/L KH2PO4 with pH 7.4 to remove urea and imidazole. After dialysis, the recombinant proteins were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting using a mouse anti-His tag antibody and injected into mice for antibody production.
For production of mouse anti-S1D antibody, 50 µg of purified S1D protein mixed with Freund’s complete adjuvant (Sigma–Aldrich, St. Louis, MO, USA) were intravenously injected into the tail of mice. As a booster, 50 µg of purified S1D with Freund’s incomplete adjuvant (Sigma–Aldrich) was subcutaneously injected into mice twice at a 10-day interval (Leenaars and Hendriksen 2005). Blood serum was collected at 3 days after the final booster and tested for antibody-binding activity with purified S1D antigen by Western blot analysis and subsequently used to detect expression of S1D protein in plants.
Agroinfiltration procedure
Nicotiana benthamiana seeds were germinated in small bottles for 2 weeks. The seedlings were transferred to bottles at one sapling per bottle. The optimal conditions for growth of N. benthamiana plants were a 16 h light/8 h dark cycle at 25 ± 0.5 °C in a greenhouse. Six-week-old plants were used for agroinfiltration.
Transient expression of plasmids pMYV717 and pMYV719 was carried out using a syringe or vacuum agroinfiltration into N. benthamiana (Leuzinger et al. 2013). For syringe agroinfiltration, A. tumefaciens harboring plasmids pMYV717, pMYV719 and pMYV508 was inoculated into 5 mL of LB medium containing kanamycin (50 mg/L) and rifampicin (50 mg/L) and incubated at 28 °C for 2 days. The cells were harvested by centrifugation and dissolved in MES infiltration buffer (10 mM MES, 10 mM MgSO4, pH 5.5, 200 µM Acetosyringone (bioWORLD, USA)) to an OD600 of 0.8. Six-week-old N. benthamiana leaves were infiltrated using a 1 mL syringe lacking a needle. The infiltrated leaves were harvested at 2–8 days after infiltration (dpi) and protein expression was assessed.
For vacuum agroinfiltration, Agrobacterium strains were inoculated into 5 mL of LB medium containing the appropriate antibiotics at 28 °C for 2 days. The inoculated bacteria were transferred into 200 mL of LB medium supplemented with appropriate antibiotics and incubated at 28 °C for 1 day. The cells were harvested by centrifugation and dissolved in 10 L of MES infiltration buffer to a final OD600 of 0.2. A 6-week-old N. benthamiana plant was placed upside down on a shelf and its leaves were submerged in MES infiltration buffer in a vacuum chamber (Fig. 2a). Vacuum at 0.1 mPa was applied for 2 min and the release valve was slowly opened to release the vacuum. The infiltrated plants were further grown in a greenhouse under 16 h continuous light per day (Fig. 2b). The infiltrated leaves were harvested at 6 dpi, lyophilized, ground and fed to mice.
Protein extraction and immunoblotting
The infiltrated leaves were harvested at 2, 4, 6 and 8 dpi. Total soluble protein was extracted from leaf tissues by grinding in liquid nitrogen with a mortar and pestle. The homogenate was suspended in Arakawa buffer extraction buffer (Arakawa et al. 1997) (1:2 w/v) (200 mM Tris–HCl, pH 8.0, 100 mM NaCl, 400 mM sucrose, 10 mM EDTA, 14 mM 2-mercaptoethanol, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 0.05 % Tween 20) and centrifuged at 13,000g for 15 min at 4 °C to remove insoluble cell debris. Protein concentration was measured by Bradford protein assay (Bio-Rad).
Aliquots of protein extracts containing 40 µg of TSP, along with prestained molecular weight markers, were separated by 10 % (non-boiled condition) or 12 % (boiled condition) using SDS-PAGE (Bio-Rad) at 120 V for 2–3 h in Tris–glycine buffer, pH 8.3 (25 mM Tris, 250 mM glycine and 0.1 % SDS). The separated protein bands were transferred onto Hybond C membranes (Amersham Pharmacia Biotech, Piscataway, NJ, USA) in transfer buffer (50 mM Tris, 40 mM glycine, and 20 % methanol) using a mini-transblot apparatus (Bio-Rad) at 130 mA for 2–3 h. To prevent non-specific antibody reactions, membranes were blocked with 10 % non-fat milk powder in TBST buffer (Tris-buffered saline with 0.05 % Tween 20) with gentle agitation on a rotary shaker at 20 rpm overnight. Membranes were incubated with a 1:5000 dilution of rabbit anti-CT antiserum (Immunology Consultants Lab, Portland, OR, USA) or mouse anti-S1D serum in TBST antibody dilution buffer containing 3.0 % non-fat dry milk. Membranes were then washed three times with TBST buffer and incubated for 2 h with a 1:5000 dilution of anti-rabbit IgG conjugated to alkaline phosphatase conjugate (S3731, Promega) or a 1:7000 dilution of anti-mouse IgG conjugated to alkaline phosphatase (S372B, Promega) in TBST buffer. Membranes were washed twice with TBST buffer and once with N buffer (100 mM Tris, pH 9.5, 5 mM MgCl2, and 100 mM NaCl). Color was developed using premixed BCIP/NBT solution (Sigma). Immunoblot membranes were subjected to densitometry using Alpha Ease FC™ software (ver. 3.3.3; AlphaInotech, San Leandro, CA, USA) to estimate protein expression levels.
PNGase F treatment
The N–glycosylated structures of S1D protein produced in plants were determined using a deglycosylation enzyme. The infiltrated leaves were ground as described above. The homogenate was suspended in denaturing extract buffer, including 0.5 % SDS and 1 % β-mercaptoethanol and subsequently centrifuged at 13,000g for 15 min at 4 °C to remove insoluble cell debris. The extracts were boiled for 10 min and chilled on ice for 5 min. The N-glycosylated sites of plant-produced protein were removed by PNGase F (ELPIS Biotech, Taejeon, Korea) in 35 µL of reaction mix, which comprised 3.5 µL of 10× reaction buffer, 1000 units of enzyme and 50 µg of protein extract for 1 h at 37 °C. The proteins were visualized using Western blot analysis as described above.
GM1-ganglioside binding activity
To examine the binding ability of the fusion proteins to gangliosides and to estimate the expression levels of CTB–S1D fusion protein in infiltrated leaves, a GM1-ganglioside enzyme-linked assay was conducted. Briefly, a 96-well microtiter plate was coated with 100 µL/well of monosialoganglioside GM1 (3.0 µg/mL) (G-7641, Sigma) dissolved in bicarbonate buffer, pH 9.6 (15 mM Na2CO3 and 35 mM NaHCO3), covered with plastic wrap and incubated overnight at 4 °C. The wells were blocked by adding 300 µL/well of 1 % bovine serum albumin (BSA) in PBS and incubated at 37 °C for 2 h, followed by three washes using PBST buffer (PBS containing 0.05 % Tween 20). The wells were loaded with serial dilutions (100 µL/well) of extracts in Arakawa protein extraction buffer and incubated for 2 h at 37 °C. The wells were washed three times with PBST, loaded with 100 µL/well of a 1:5000 dilution of rabbit anti-CT primary antibody or mouse anti-S1D antibody, and incubated for 2 h at 37 °C, followed by washing three times with PBST buffer. The plate was then incubated with 100 µL/well of secondary antibody, a 1:5000 dilution of alkaline phosphatase-conjugated anti-rabbit IgG (A-2556, Sigma) or a 1:7000 dilution of anti-mouse IgG conjugated with alkaline phosphatase (S372B, Promega) for 2 h at 37 °C and washed three times with PBST buffer. The plate was finally incubated for 15 min at room temperature with 100 µL/well of phosphatase substrate (S0942, Sigma). Optical density was measured at 405 nm wavelength in an ELISA reader (MRA-006; Packard Instrument Co., Meriden, CT, USA).
Oral immunization with lyophilized powder of infiltrated leaves
The CTB–S1D fusion protein, which was expressed at a high level in infiltrated leaves, was selected for mouse oral gavage. Female BALB/c mice were purchased from Orientbio, Inc. (Seongnam, Korea) and randomly divided into four groups of five mice each (Fig. 6a). Experimental mice were administered the powder through oral gavage every week for 6 weeks (Huy et al. 2012), as shown in Fig. 6b. For feeding immunization, the mice were fasted 8 h before gavage with 2.0 mL of PBS buffer pH 7.0 containing lyophilized leaf powder, which harbored 100 µg of CTB–S1D fusion protein, or with bacterial cholera toxin—bCT (Sigma) or rice callus-expressed mutant cholera toxin 61 F (Arg to Phe)—rCTX and wild-type (WT) N. benthamiana leaves. Blood and fecal samples were collected before feeding and 3 days after feeding at weeks 4 and 6 (Jespersgaard et al. 1999). The collected blood samples were stored at room temperature for 1 h, chilled on ice for 1 h and centrifuged at 13,000 rpm for 10 min. Sera were collected and stored at −70 °C for antibody assays. Fecal samples (200 mg) were dissolved in 1 mL of PBS buffer containing 0.01 % sodium azide. Insoluble materials were removed by centrifugation at 13,000 rpm for 10 min. Fecal extracts were also stored at −70 °C for analysis of specific secretory IgA (sIgA).
Detection of specific antibody responses in sera and fecal extracts
For analysis of specific systemic antigen-specific IgG and sIgA, an ELISA was conducted according to a described protocol (Kim et al. 2010) with minor modification. Briefly, the ELISA plates were coated with 100 µL/well of antigen (80 ng/well) dissolved in bicarbonate buffer (15 mM Na2CO3, 25 mM NaHCO3, pH 9.6) covered with plastic wrap and incubated overnight at 4 °C. The wells were washed thrice with PBST (PBS plus 0.05 % Tween 20), blocked by addition of 300 µL per well of 1 % BSA in PBS buffer, and incubated at 37 °C for 2 h, followed by three washes with PBST. The test sera and fecal extracts were diluted in PBS buffer at 1:100 and 1:4, respectively, which was loaded into the antigen-coated plates and incubated at 37 °C for 2 h. The plates were washed thrice with PBST, and then incubated with 100 µL per well of a 1:7000 dilution of anti-mouse IgG (A-3688, Sigma) or anti-mouse IgA (A-4937, Sigma) conjugated to alkaline phosphatase for 2 h at 37 °C and washed thrice with PBST. The plates were developed by addition of 100 µL per well of alkaline phosphatase buffer (10 % (v/v) diethanolamine, 0.1 % MgCl2, 0.02 % sodium azide, pH 9.8) plus one tablet of phosphate substrate (S0942-100TAB, Sigma) for 30 min at room temperature in the dark. The plates were read at 405 nm wavelength in an ELISA reader (Packard Instrument Co.).
Data analysis
The data were analyzed using Excel 2007 software (Microsoft Corp, Redmond, WA, USA) and expressed as means ± SD of at least three independent experiments. The differences in the levels of total IgG and sIgA antibodies were evaluated by one-way ANOVA. Statistical significance was determined using SPSS® for Windows software (ver. 21.0; SPSS Inc., Chicago, IL, USA). P < 0.05 was considered to indicate statistical significance.
Results
Construction of plant expression vectors
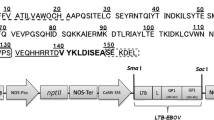
The S1D gene alone was cloned into a plant expression vector, yielding plasmid pMYV717 (Fig. 1a), under the regulation of the duplicated CaMV 35S promoter, CTB signal peptide and terminator of nopaline synthase. To enhance the expression level, the Kozak sequence (Kang et al. 2004) was added upstream of the start codon and the ER retention signal (SEKDEL) at the C-terminus of the S1D gene.

T-DNA constructs used for agroinfiltration. Binary vectors for transient expression of S1D protein (a), CTB–S1D fusion protein (b) and p19 protein (c). LB left border of T-DNA; RB right border of T-DNA; S1D S1D epitope; CTB cholera toxin B subunit; dp35S duplicated CaMV 35S promoter; Nos-T terminator of nopaline synthase, Nos-P promoter of nopaline synthase; sp CTB signal peptide; Kozak Kozak consensus sequence; SEKDEL ER retention signal; NPT II neomycin phosphotransferase II kanamycin resistance gene; p19 suppressor gene of the p19 protein of tomato bushy stunt virus
To express the CTB–S1D fusion gene, the S1D gene was inserted into a plant expression vector containing the duplicated CaMV 35S promoter, the CTB gene and the nos terminator, yielding plasmid pMYV719 (Fig. 1b). Plasmid pMYV508 contains the p19 gene, which encodes the suppressor of gene silencing under the control of the duplicated CaMV 35S promoter and the nos terminator (Fig. 1c). The plasmids pMYV717, pMYV719 and pMYV508 were transformed into A. tumefaciens LBA4404 for agroinfiltration into N. benthamiana leaves. The conditions for vacuum infiltration with intact leaves were optimized in our laboratory (Fig. 2). Vacuum agroinfiltration for production of transiently expressed proteins is highly scalable.

Vacuum agroinfiltration of N. benthamiana plants with Agrobacterium tumefaciens. a The vacuum chamber was connected to a vacuum pump. The entire leaf system of a 6-week-old plant was placed upside down with leaves submerged in MES infiltration buffer in a vacuum chamber. b Left and right, infiltrated plant and plant prepared for vacuum infiltration, respectively
Western blot analysis of transient S1D expression
Agrobacterium tumefaciens LBA4404 harboring the plasmid pMYV717 was co-infiltrated without or with the p19 construct (pMYV508) (Fig. 3a) into N. benthamiana leaves. The infiltrated leaves were harvested at 2, 4, 6 and 8 dpi. Immunoblot analysis was conducted to detect S1D protein in infiltrated leaves. The S1D protein was detected with a molecular weight of ~34 kDa (Fig. 3a, b) in comparison with the bacterial S1D positive control, which was observed at ~17 kDa (Fig. 3a–f). No background signal bands corresponding to the S1D protein were detected in protein extracts from leaves infiltrated with p19 at 4 dpi, as a negative control. Leaves infiltrated with S1D without p19 showed high-density bands at 2 dpi, whereas the S1D levels in the presence of p19 (+p19) were significantly increased, and peaked at 4 dpi (Fig. 3a).

Western blot analyses of transiently expressed S1D protein and CTB–S1D fusion protein in agroinfiltrated N. benthamiana leaves. Transient expression of plasmid pMYV717 containing the S1D gene (a) co-infiltrated with (+ p19) or without plasmid pMYV508 (p19). Infiltrated leaves were sampled at 2, 4, 6 and 8 dpi. Protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed using a mouse anti-S1D primary antibody. Analysis of N-glycosylation of S1D protein with PNGase F(+) (b). Transient expression of the CTB–S1D fusion protein in agroinfiltrated N. benthamiana leaves. N. benthamiana leaves were co-infiltrated with plasmid pMYV719 with or without p19 and sampled at 2, 4, 6 and 8 dpi. Protein extracts were separated by SDS-PAGE and analyzed after boiling (e, f) or not (c, d) using an anti-cholera toxin (c, e) or anti-S1D (d, f) primary antibody. Lane M, prestained protein ladder (Fermentas, Hanover, MD, USA); bS1D, S1D antigen expressed in E. coli; bCTB, commercial bacterial cholera toxin B subunit (Sigma); p19-4, 4 dpi leaves infiltrated with plasmid pMYV508 used as a negative control. P and M indicate the pentamer and monomer of CTB–S1D fusion protein, respectively
The S1D protein has seven potential N-glycosylation sites. To analyze N-glycosylation status, protein extracts were treated with PNGase F (Fig. 3b) to deglycosylate the glycan(s) from the core peptide in the denatured protein. The deglycosylated S1D protein band was lower than the untreated bands and migrated in a manner similar to the bacterial S1D protein (Fig. 3b). This indicates that S1D protein underwent glycosylation in infiltrated leaves.
Western blot analysis of CTB–S1D transient expression
The CTB–S1D fusion protein was detected in both boiled and unboiled protein extracts from infiltrated leaves using anti-CT and anti-S1D antibodies. No signal band corresponding to the CTB–S1D protein was detected in protein extracts from leaves infiltrated with p19 at 4 dpi, as a negative control. In the unboiled condition, assembly of CTB–S1D fusion protein into oligomeric structures resembling pentamers with a molecular weight of over 170 kDa was detected using anti-CT and anti-S1D antibodies (Fig. 3c, d), in comparison with the bacterial CTB positive control, which was observed at ~43 kDa (Fig. 3c). A 17 kDa band was observed for the bacterial S1D protein (Fig. 3d, f). When protein extracts were boiled for 5 min, the oligomeric fusion protein dissociated into ~ 51 kDa monomers and was detected by both anti-CT and anti-S1D antibodies (Fig. 3e, f). The monomer of the CTB protein was detected at ~12 kDa (Fig. 3e). The molecular weight of the monomer of CTB–S1D based on in silico analysis is ~30 kDa. The high molecular weight of plant-derived CTB–S1D (~51 kDa) may be due to glycosylation of CTB (2 potential N-glycosylation sites) and S1D (seven potential N-glycosylation sites, Fig. 3e, f), resulting in retardation of migration. The densities of CTB–S1D fusion proteins were significantly increased in the presence (+p19) of p19 (Fig. 3c–f).
GM1-ganglioside binding activity
The biological function of the CTB–S1D fusion protein produced in infiltrated leaves was determined by GM1-ELISA. The GM1-ganglioside–binding capacity of CTB–S1D fusion protein was determined using anti-CT (Fig. 4a) and anti-S1D (Fig. 4b) primary antibodies. These results indicated that the monomeric CTB–S1D fusion proteins were properly assembled into a biologically functional form and that S1D was conserved in the fusion protein.

GM1-ELISA of CTB–S1D fusion protein produced in infiltrated N. benthamiana leaves. The biological activity of CTB–S1D fusion protein with the intestinal membrane GM1-ganglioside receptor was confirmed using an anti-cholera toxin antibody (a) and anti-S1D antibody (b)
Expression levels of S1D and CTB–S1D fusion protein
The immunoblot membranes were subjected to densitometry to estimate the level of S1D protein using the Alpha Ease FC™ software. Predetermined protein concentrations (20, 40 and 80 ng) were used to generate a standard curve. Protein expression levels were estimated based on a time-course analysis. The expression levels of transient proteins were expressed as percentages of total soluble protein (% of TSP). The highest expression level of S1D was found at 2 dpi, and reached 0.04 % of TSP (Fig. 5a). In the presence of p19, expression levels significantly increased to 0.15 % of TSP at 4 dpi (Fig. 5a).

The expression levels of S1D protein (a) and CTB–S1D fusion protein (b) with or without p19 in infiltrated N. benthamiana leaves at 2, 4, 6 and 8 dpi. Expression levels are presented as percentages of TSP. Error bars represent standard deviations
GM1-ELISA was employed to evaluate the expression levels of the assembled CTB–S1D fusion protein. The highest amount of assembled CTB–S1D fusion protein was 0.07 % of TSP at 4 dpi, and decreased rapidly thereafter. The CTB–S1D expression levels in the presence of p19 reached 0.49 % of TSP (Fig. 5b), and rapidly increased from 2 to 4 dpi, peaking at 6 dpi. This result indicates that p19 enhances the expression levels and assembly of CTB–S1D fusion protein into biologically functional molecules.
Detection of specific serum IgG and fecal IgA responses
Mice were orally immunized with WT or transient leaf powder containing CTB–S1D protein (CTB–S1D) alone, CTB–S1D protein with rice-expressed mutant CT61F (CTB–S1D + rCTX) and with bacterial CT (CTB–S1D + bCT) adjuvants on six separate occasions using appropriate doses of antigen and adjuvant (Fig. 6). To determine the levels of anti-CTB and anti-S1D antibodies, sera and fecal extracts were subjected to ELISA. The anti-CTB and anti-S1D IgG and sIgA were significantly higher in all experimental groups at 4 weeks of feeding, and peaked at 6 weeks of feeding (Fig. 7). As expected, antibodies were not detected in the control group. Anti-CTB and anti-S1D IgG levels were significantly increased in mice administered CTB–S1D + bCT and CTB–S1D + rCTX in comparison with mice given CTB–S1D at 6 weeks (Fig. 7a, b). Moreover, anti-CTB and anti-S1D sIgA levels were significantly higher in mice given CTB–S1D + bCT compared with those administered CTB–S1D and CTB–S1D + rCTX at 6 weeks (Fig. 7c, d). Oral immunization with transient expression CTB–S1D fusion protein induced significantly higher anti-CTB, anti-S1D IgG and sIgA levels compared with the control group (Fig. 7).

Mouse feeding strategy. a Female BALB/c mice were randomly divided into four groups of five mice each. Wild-type (WT) and infiltrated N. benthamiana leaves were lyophilized and ground. Mice in the experimental groups were orally immunized with infiltrated leaf powder containing 100 µg of CTB–S1D fusion protein, 100 µg of CTB–S1D fusion protein with 5 µg of rice calli-expressed CT61F (rCTX) and 5 µg of bacterial cholera toxin (bCT) in 2 mL of PBS buffer. The mice in the negative control group were fed WT leaf powder. Blood and fecal samples were collected 3 days after feeding at weeks 4 and 6 as well as 3 days before starting feeding (b)

Induction of specific antibodies in mice. Serum IgG and sIgA against CTB antigen (a–c) and S1D antigen (b–d) in mice at weeks 0, 4, and 6. Mouse sera and fecal samples were diluted 100- and fourfold, respectively, and used in ELISAs. Results are mean OD (405 nm) values of five immunized mice. Error bars represent standard deviations. Any two means in a column with the same letter in common are not significantly (P ≤ 0.05) different according to Tukey’s test. Different letters within a column indicate significant differences (P ≤ 0.05) according to Tukey’s test
Discussion
PEDV causes acute enteritis characterized by vomiting and watery, fetid diarrhea in affected pigs of all ages. The mortality rate in piglets is 90–95 %, leading to significant economic losses (Debouck and Pensaert 1980; Steinrigl et al. 2015). PED was reported as a re-emergent disease in the United States and neighboring countries, such as Canada and Central American, European and Asian countries (Beam et al. 2015; Li et al. 2012; Steinrigl et al. 2015). In late 2013, a severe outbreak of PEDV infection occurred in Korea, causing economic losses to the pork industry (Lee et al. 2015). Therefore, an effective vaccine to protect pigs against PEDV infection is required. The neutralizing COE epitope (aa 499–638) in the spike glycoprotein was identified (Chang et al. 2002). Based on optimized codon usage in plant, expression of the synthetic COE gene was fivefold higher than that of the native gene in transgenic tobacco plants (Kang et al. 2005). To enhance the immune responses, the synthetic COE gene was fused with a mucosal adjuvant and carrier, LTB and CTB, respectively, and with the M cell-target peptide ligand Co1, and expressed in various plant species including tobacco (Kang et al. 2005), lettuce (Huy et al. 2009, 2011) and rice callus (Huy et al. 2012). Oral immunization with COE-Co1 fusion protein expressed in transgenic rice callus induced systemic and mucosal immune responses against the COE antigen (Huy et al. 2012). Recently, S1D (aa 636–789) has been seen as an alternative neutralizing epitope domain in the spike protein of PEDV (Sun et al. 2008). In this report, the S1D epitope alone or fusion protein was expressed in infiltrated N. benthamiana by agroinfiltration. The immunogenicity of the transiently expressed proteins was investigated to develop an edible vaccine against PEDV.
Plant cells have been widely used to express recombinant vaccine antigens. Recombinant proteins can be expressed in plants by creating stable transgenic plants or by performing transient expression. Transient expression offers several advantages: accumulation of high levels of protein, short timeline of protein production, scalability and elimination of transgene outflow (Chen et al. 2013). To optimize S1D expression, the gene was added to various expression vectors, including the Agrobacterium vector pTRAC (Maclean et al. 2007), a viral pro-vector (Marillonnet et al. 2004) and a binary vector. The target protein was expressed at a high level using the binary vector. However, the level of protein production was low (Figs. 3a, 4a), as it was limited by the PTGS factor. The p19 protein of TBSV prevents the onset of PTGS in infiltrated leaves and enhances the levels of transiently co-expressed proteins (Mohammadzadeh et al. 2015; Voinnet et al. 2003). The S1D and CTB–S1D protein levels were enhanced from fourfold to sevenfold in the presence of p19 (Figs. 3, 4), respectively. Moreover, accumulation of the CTB–S1D fusion protein was fourfold greater than that of the S1D protein (Fig. 4). The CTB portion plays a role in enhancing protein accumulation and as a mucosal adjuvant and carrier. In previous studies, the expression level of synthetic COE-Co1 was 0.083 % of TSP in transgenic rice under the control of the strong RAmy3D promoter at day 3 after induction in sucrose starvation medium (Huy et al. 2012). The highest expression level of CTB-COE fusion protein was 0.0065 % of TSP in transgenic lettuce plants (Huy et al. 2011). By performing transient expression in the presence of p19, the expression levels of S1D protein and CTB–S1D fusion protein were 2- and 75-fold higher than those of COE-Co1 and CTB-COE in transgenic plants. The increased protein expression levels might result in elicitation of strong immune responses and prevent induction of immune tolerance by plant-based oral vaccines. The binding of functional CTB–S1D fusion protein to its biological receptor, GM1-ganglioside, was confirmed by GM1-ELISA with anti-CT and anti-S1D antibodies (Fig. 4). These results indicated that the S1D protein of the CTB–S1D fusion protein was assembled into functional oligomeric structures and retained its antigenicity.
To investigate the immunogenicity of the transiently expressed protein, the CTB–S1D fusion gene was transiently expressed in N. benthamiana leaves by vacuum agroinfiltration (Fig. 2). The infiltrated leaves were lyophilized and ground, and the expression of assembled CTB–S1D fusion protein was confirmed before oral administration to female BALB/c mice for 6 weeks at a 1-week interval (Fig. 6b). Mice given the CTB–S1D fusion protein exhibited CTB- and S1D-specific systemic IgG at week 4. The IgG and sIgA levels were significantly increased at week 6 (Fig. 7a-d). CT acts not only as a mucosal immunogen and mucosal adjuvant but also as induces immune responses to unrelated nasally and orally co-administered antigens (Elson and Ealding 1984; Isaka et al. 2004). The CT61F mutant, which lacks ADP-ribosylation activity but retains the mucosal adjuvant properties of native CT, was expressed in rice callus (unpublished paper). To enhance the immune responses, transiently expressed CTB–S1D was co-gavaged with bCT and rCTX into mice at appropriate doses (Fig. 6a). As expected, anti-CTB and anti-S1D Ab levels, in mice given CTB–S1D with bCT and with rCTX at 6 weeks (Fig. 7a, b), were significantly higher than those of mice fed CTB–S1D only. sIgA is the predominant antibody class in external secretions and the major humoral defense factor that prevents colonization of mucosal surfaces by infective agents, such as bacteria and viruses (Haan et al. 1995; Woof and Mestecky 2005). This sIgA may thus prevent PED. The levels of S1D-specific sIgA responses were significantly greater than those in the control group. The anti-S1D antibody levels in mice given CTB–S1D and bCT were significantly increased in comparison with those fed CTB–S1D only (Fig. 7d). The transient expression of CTB–S1D fusion protein thus induced S1D-specific systemic and mucosal immune responses following oral administration to mice. bCT and rCTX enhanced S1D-specific antibody responses when orally co-administered with CTB–S1D protein.
In conclusion, the S1D gene alone, and that fused with the CTB gene, were expressed in infiltrated N. benthamiana leaves. Oral immunization of mice with CTB–S1D fusion protein elicited systemic and mucosal immune responses. The CTB–S1D fusion protein could be used to produce a plant-based vaccine against PEDV.
References
Arakawa T, Chong DK, Merritt JL, Langridge WH (1997) Expression of cholera toxin B subunit oligomers in transgenic potato plants. Transgenic Res 6(6):403–413
Beam A, Goede D, Fox A, McCool MJ, Wall G, Haley C, Morrison R (2015) A porcine epidemic diarrhea virus outbreak in one geographic region of the united states: descriptive epidemiology and investigation of the possibility of airborne virus spread. Plos One 10(12):e0144818. doi:10.1371/journal.pone.0144818
Chang SH, Bae JL, Kang TJ, Kim J, Chung GH, Lim CW, Laude H, Yang MS, Jang YS (2002) Identification of the epitope region capable of inducing neutralizing antibodies against the porcine epidemic diarrhea virus. Mol Cells 14(2):295–299
Chasey D, Cartwright SF (1978) Virus-like particles associated with porcine epidemic diarrhoea. Res Vet Sci 25(2):255–256
Chen Q, Lai H, Hurtado J, Stahnke J, Leuzinger K, Dent M (2013) Agroinfiltration as an effective and scalable strategy of gene delivery for production of pharmaceutical proteins. Adv Tech Biol Med 1(1):103 doi:10.4172/atbm.1000103
Debouck P, Pensaert M (1980) Experimental infection of pigs with a new porcine enteric coronavirus, CV 777. Am J Vet Res 41(2):219–223
Duarte M, Laude H (1994) Sequence of the spike protein of the porcine epidemic diarrhoea virus. J Gen Virol 75(5):1195–1200 doi:10.1099/0022-1317-75-5-1195
Ducatelle R, Coussement W, Charlier G, Debouck P, Hoorens J (1981) Three-dimensional sequential study of the intestinal surface in experimental porcine CV 777 coronavirus enteritis. Zentralbl Vet B 28(6):483–493
Elson CO, Ealding W (1984) Cholera toxin feeding did not induce oral tolerance in mice and abrogated oral tolerance to an unrelated protein antigen. J Immunol 133(6):2892–2897
Follis KE, York J, Nunberg JH (2006) Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology 350(2):358–369 doi:10.1016/j.virol.2006.02.003
Haan Ad, Renegar KB, Small PA Jr, Wilschut J (1995) Induction of a secretory IgA response in the murine female urogenital tract by immunization of the lungs with liposome-supplemented viral subunit antigen. Vaccine 13(7):613–616. doi:10.1016/0264-410X(94)00062-R
Hernández M, Rosas G, Cervantes J, Fragoso G, Rosales-Mendoza S, Sciutto E (2014) Transgenic plants: a 5-year update on oral antipathogen vaccine development. Expert Rev Vaccines 13(12):1523–1536. doi:10.1586/14760584.2014.953064
Horsch RB, Fry JE, Hoffmann NL, Eichholtz D, Rogers SG, Fraley RT (1985) A simple and general method for transferring genes into plants. Science 227(4691):1229–1231. doi:10.1126/science.227.4691.1229
Huy NX, Kim YS, Jun SC, Jin Z, Park SM, Yang MS, Kim TG (2009) Production of a heat-labile enterotoxin B subunit-porcine epidemic diarrhea virus-neutralizing epitope fusion protein in transgenic lettuce (Lactuca sativa). Biotechnol Bioprocess Eng 14(6):731–737
Huy NX, Yang MS, Kim TG (2011) Expression of a cholera toxin B subunit-neutralizing epitope of the porcine epidemic diarrhea virus fusion gene in transgenic lettuce (Lactuca sativa L.) Mol Biotechnol 48(3):201–209. doi:10.1007/s12033-010-9359-1
Huy NX, Kim SH, Yang MS, Kim TG (2012) Immunogenicity of a neutralizing epitope from porcine epidemic diarrhea virus: M cell targeting ligand fusion protein expressed in transgenic rice calli. Plant Cell Rep 31(10):1933–1942. doi:10.1007/s00299-012-1306-0
Isaka M, Komiya T, Takahashi M, Yasuda Y, Taniguchi T, Zhao Y, Matano K, Matsui H, Maeyama JI, Morokuma K, Ohkuma K, Goto N, Tochikubo K (2004) Recombinant cholera toxin B subunit (rCTB) as a mucosal adjuvant enhances induction of diphtheria and tetanus antitoxin antibodies in mice by intranasal administration with diphtheria–pertussis–tetanus (DPT) combination vaccine. Vaccine 22(23–24):3061–3068. doi:10.1016/j.vaccine.2004.02.019
Jespersgaard C, Hajishengallis G, Greenway TE, Smith DJ, Russell MW, Michalek SM (1999) Functional and immunogenic characterization of two cloned regions of Streptococcus mutans glucosyltransferase I. Infect Immun 67(2):810–816
Kang T-J, Loc N-H, Jang M-O, Yang M-S (2004) Modification of the cholera toxin B subunit coding sequence to enhance expression in plants. Mol Breed 13(2):143–153. doi:10.1023/B:MOLB.0000018762.27841.7a
Kang T-J, Kim Y-S, Jang Y-S, Yang M-S (2005) Expression of the synthetic neutralizing epitope gene of porcine epidemic diarrhea virus in tobacco plants without nicotine. Vaccine 23(17–18):2294–2297. doi:10.1016/j.vaccine.2005.01.027
Kim TG, Huy NX, Kim MY, Jeong DK, Jang YS, Yang MS, Langridge WH, Lee JY (2009) Immunogenicity of a cholera toxin B subunit Porphyromonas gingivalis fimbrial antigen fusion protein expressed in E. coli. Mol Biotechnol 41(2):157–164 doi:10.1007/s12033-008-9102-3
Kim TG, Kim BG, Kim MY, Choi JK, Jung ES, Yang MS (2010) Expression and immunogenicity of enterotoxigenic Escherichia coli heat-labile toxin B subunit in transgenic rice callus. Mol Biotechnol 44(1):14–21 doi:10.1007/s12033-009-9200-x
Kocherhans R, Bridgen A, Ackermann M, Tobler K (2001) Completion of the porcine epidemic diarrhoea coronavirus (PEDV) genome sequence. Virus Genes 23(2):137–144
Lal P, Ramachandran VG, Goyal R, Sharma R (2007) Edible vaccines: current status and future. Indian J Med Microbiol 25(2):93–102
Lee S, Lee C (2014) Outbreak-related porcine epidemic diarrhea virus strains similar to US strains, South Korea, 2013. Emerg Infect Dis 20(7):1223–1226. doi:10.3201/eid2007.140294
Lee S, Kim Y, Lee C (2015) Isolation and characterization of a Korean porcine epidemic diarrhea virus strain KNU-141112. Virus Res 208:215–224. doi:10.1016/j.virusres.2015.07.010
Leenaars M, Hendriksen CFM (2005) Critical steps in the production of polyclonal and monoclonal antibodies: evaluation and recommendations. ILAR J 46(3):269–279. doi:10.1093/ilar.46.3.269
Leuzinger K, Dent M, Hurtado J, Stahnke J, Lai H, Zhou X, Chen Q (2013) Efficient agroinfiltration of plants for high-level transient expression of recombinant proteins. J Vis Exp doi:10.3791/50521
Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q (2012) New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18(8):1350–1353. doi:10.3201/eid1808.120002
Maclean J, Koekemoer M, Olivier AJ, Stewart D, Hitzeroth II, Rademacher T, Fischer R, Williamson AL, Rybicki EP (2007) Optimization of human papillomavirus type 16 (HPV-16) L1 expression in plants: comparison of the suitability of different HPV-16 L1 gene variants and different cell-compartment localization. J Gen Virol 88(5):1460–1469. doi:10.1099/vir.0.82718-0
Marillonnet S, Giritch A, Gils M, Kandzia R, Klimyuk V, Gleba Y (2004) In planta engineering of viral RNA replicons: efficient assembly by recombination of DNA modules delivered by Agrobacterium. Proc Natl Acad Sci U S A 101(18):6852–6857. doi:10.1073/pnas.0400149101
Mohammadzadeh S, Roohvand F, Memarnejadian A, Jafari A, Ajdary S, Salmanian A-H, Ehsani P (2015) Co-expression of hepatitis C virus polytope–HBsAg and p19-silencing suppressor protein in tobacco leaves. Pharm Biol:1–9 doi:10.3109/13880209.2015.1048371
Munro S, Pelham HR (1987) A C-terminal signal prevents secretion of luminal ER proteins. Cell 48(5):899–907
Pensaert MB, de Bouck P (1978) A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58(3):243–247
Poland GA, Murray D, Bonilla-Guerrero R (2002) New vaccine development. BMJ. Br Med J 324(7349):1315–1319
Puranaveja S, Poolperm P, Lertwatcharasarakul P, Kesdaengsakonwut S, Boonsoongnern A, Urairong K, Kitikoon P, Choojai P, Kedkovid R, Teankum K, Thanawongnuwech R (2009) Chinese-like strain of porcine epidemic diarrhea virus, Thailand. Emerg Infect Dis 15(7):1112–1115. doi:10.3201/eid1507.081256
Sala F, Manuela Rigano M, Barbante A, Basso B, Walmsley AM, Castiglione S (2003) Vaccine antigen production in transgenic plants: strategies, gene constructs and perspectives. Vaccine 21(7–8):803–808. doi:10.1016/S0264-410X(02)00603-5
Steinrigl A, Revilla Fernández S, Stoiber F, Pikalo J, Sattler T, Schmoll F (2015) First detection, clinical presentation and phylogenetic characterization of porcine epidemic diarrhea virus in Austria. BMC Vet Res 11:310. doi:10.1186/s12917-015-0624-1
Stevenson GW, Hoang H, Schwartz KJ, Burrough ER, Sun D, Madson D, Cooper VL, Pillatzki A, Gauger P, Schmitt BJ, Koster LG, Killian ML, Yoon KJ (2013) Emergence of porcine epidemic diarrhea virus in the United States: clinical signs, lesions, and viral genomic sequences. J Vet Diagn Investig 25(5):649–654 doi:10.1177/1040638713501675
Strugnell R, Zepp F, Cunningham A, Tantawichien T (2011) Vaccine antigens. Perspect Vaccinol 1(1):61–88 doi:10.1016/j.pervac.2011.05.003
Sun D, Feng L, Shi H, Chen J, Cui X, Chen H, Liu S, Tong Y, Wang Y, Tong G (2008) Identification of two novel B cell epitopes on porcine epidemic diarrhea virus spike protein. Vet Microbiol 131(1–2):73–81. doi:10.1016/j.vetmic.2008.02.022
Takahashi K, Okada K, Ohshima K (1983) An outbreak of swine diarrhea of a new-type associated with coronavirus-like particles in Japan. Nihon Juigaku Zasshi 45(6):829–832
Turgeon DC, Morin M, Jolette J, Higgins R, Marsolais G, DiFranco E (1980) Coronavirus-like particles associated with diarrhea in baby pigs in Quebec. Can Vet J 21(3):100
Voinnet O, Rivas S, Mestre P, Baulcombe D (2003) An enhanced transient expression system in plants based on suppression of gene silencing by the p19 protein of tomato bushy stunt virus. Plant J 33(5):949–956
Woof JM, Mestecky J (2005) Mucosal immunoglobulins. Immunol Rev 206:64–82. doi:10.1111/j.0105-2896.2005.00290.x
Acknowledgments
This research was supported by the Agriculture, Food and Rural Affairs Research Center Support Program, Ministry of Agriculture, Food and Rural Affairs, and Nguyen-Quang-Duc Tien was supported by the BK21 plus program, Republic of Korea.
Author contributions
Nguyen-Xuan Huy, Nguyen-Quang-Duc Tien and Moon-Sik Yang conceived, designed and performed the overall study. Mi-Young Kim generated the transformed rice callus expressing mutant cholera toxin 61F. Tae-Geum Kim and Yong-Suk Jang designed the mouse experiment. Nguyen-Xuan Huy wrote the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Nguyen-Xuan Huy and Nguyen-Quang-Duc Tien have contributed equally to this study.
Rights and permissions
About this article

Cite this article
Huy, NX., Tien, NQD., Kim, MY. et al. Immunogenicity of an S1D epitope from porcine epidemic diarrhea virus and cholera toxin B subunit fusion protein transiently expressed in infiltrated Nicotiana benthamiana leaves. Plant Cell Tiss Organ Cult 127, 369–380 (2016). https://doi.org/10.1007/s11240-016-1059-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-016-1059-5