
Abstract
Infection with Human papillomaviruses (HPVs) is linked to cervical cancer, which is one of the most common cancers found among women worldwide. Despite preventive immunization, a therapeutic vaccine, targeting infected individuals, is required. Vaccine strategies for treatment of HPV—induced cervical cancer are based on E7 and E6 oncoproteins. In this work we report the transient expression of chimeric particles containing the E6 oncoprotein from Human papillomavirus type 16 (HPV-16) in plants. We fused a mutagenized coding sequence of the E6 oncoprotein (E6GT) with the coding sequence of Potato virus X coat protein (PVX CP) both with the 5′- and 3′-terminus using linkers of different length (0, 4 or 15 amino acids). The expression in E. coli was performed to assess the characteristics of the recombinant protein prior to plant expression. The yield and immunological reactivity of the expressed proteins were screened with anti-PVX CP and E6 antibodies. The highest yields of chimeric particles were observed in the transgenic N. benthamiana expressing Potato virus A HC-Pro protein and the Tobacco mosaic virus movement protein. When inoculated on host plants, these recombinant viruses were not able to spread systemically. The obtained results revealed the new relations for design of expression cassettes for plant-based vaccine production.
Similar content being viewed by others


Introduction
Human papillomaviruses (HPVs) cause cervical carcinoma and are associated with several other human anogenital cancers (Ganguly and Parihar 2009). They are small viruses with a double-stranded circular DNA, which replicates in the nucleus of infected cells. Among the HPV proteins, the early proteins E6 and E7 attract most attention as they transform and immortalize cells (Ganguly and Parihar 2009; McLaughlin-Drubin and Munger 2009; Hsu et al. 2011).
The availability of prophylactic vaccines against HPVs (based on HPV-16, -18, -6, and -11 L1 structural proteins assembled in virus-like particles—VLPs) represents a milestone in the prevention of this infection (Koutsky et al. 2002; Villa et al. 2005; Harper et al. 2006), establishing the base for a significant reduction of the rate of cervical cancer in future. Despite this preventive immunization, a therapeutic vaccine, targeting already infected individuals, is also required. Many candidate vaccines with therapeutic potential are currently tested in ongoing trials; however, there is low expectation that any of the current therapeutic vaccines will have a substantial public health impact in the near future (Ma et al. 2010). Several therapeutic HPV-specific vaccines based on E7 oncoprotein, which is the primary target of therapeutic vaccines, designed to treat later stages of the disease, have been tested in animal models and some have advanced into phase II and III clinical trials (Münger et al. 1989; Billich 2003; Frazer 2004). There is an increasing demand for gene expression systems that allow for high levels of foreign protein production in plants, whether transgenically, transiently or by means of a virus vector (Scholthof et al. 2002; Canizares et al. 2005; Faye and Gomord 2010; Anwar et al. 2011; Kim et al. 2011; Martínez-González et al. 2011).
Vectors derived from Potato virus X (PVX) are widely used to rapidly express high levels of recombinant proteins e.g. vaccines in plants. PVX coat protein (PVX CP) fusion with its ability to form virus–like particles is a suitable carrier for display of antigens accessible to the immune system.
There were numerous attempts to produce vaccines using N-terminal fusions of antigens with PVX CP which were able to form virus-like particles (Marusic et al. 2001; Lico et al. 2006, 2009; Uhde-Holzem et al. 2007; Marconi et al. 2009; Morgenfeld et al. 2009). There is also number of works regarding expression of HPV antigens in plants (Franconi et al. 2002; Biemelt et al. 2003; Varsani et al. 2003; Liu et al. 2005; Maclean et al. 2007; Fernández-San Millán et al. 2008; De la Rosa et al. 2009). However, successful expression of the E6 oncoprotein in plants has not yet been reported. Therefore we designed a transient expression system for this antigen as N- or C-terminal PVX CP fusions first in E. coli to determine their properties, and then for expression in plants.
The mutagenized HPV16 E6 protein (E6GT) has been proposed as a safer candidate than the wild type E6 for vaccine development. The introduction of double mutation into the E6 protein led to the loss of the ability to induce the p53 degradation (Polakova et al. 2010).
We previously focused on the development of transient expression systems based on plant viral vectors for production of different epitopes from Human papillomavirus type 16 (HPV-16) minor coat protein L2 and oncoprotein E7 or on the expression of mutagenized HPV-16 E7 oncoprotein in plants (Cerovska et al. 2008, 2012; Hoffmeisterova et al. 2012; Plchova et al. 2011).
To display E6GT on the surface of PVX CP VLPs, we used a transient expression system previously designed for expression of the E7ggg protein (Plchova et al. 2011). This system allows to express different full-length antigens fused either to the N- or C-terminus of PVX CP using linkers of different lengths to find the optimal expression. We studied the impact of the topology of the fusion and the effect of linker length on expression levels both in Escherichia coli and Nicotiana benthamiana.
Materials and methods
Cloning of fusion constructs
To avoid safety concerns associated with the administration of an oncogene in humans, we used an attenuated HPV-16 E6 gene, termed E6GT, that was demonstrated to prevent E6-mediated p53 degradation: it carries two amino acid substitutions, C70G and I135T (Polakova et al. 2010).
E6GT was amplified by PCR from vector pBSC-E6GT using either G9-R and G9-U or G9-P and G9-O primers, respectively (Table 1; Fig. 1). The first PCR product was inserted into the PVX CP-BH/pMPM-A4Ω plasmid (Plchova et al. 2011) using the XbaI and BglII restriction sites to create the final expression plasmid E6GT-PVX CP/pMPM-A4Ω carrying the E6GT fused to the 5′-terminus of PVX CP. The second PCR product was cloned into the PVX CP-NB/pMPM-A4Ω plasmid (Plchova et al. 2011) using the BglII and SalI restriction sites to produce the final PVX CP-E6GT/pMPM-A4Ω expression plasmid carrying the E6GT fused to the 3′-terminus of PVX CP.

Schematic illustration of the genetic constructs in bacterial expression vector pMPM-A4Ω. Restriction endonuclease sites and positions of oligonucleotides (black arrows) used for preparation of chimeric constructs are indicated. E6GT—mutated form of E6 from HPV16; L4—4 amino acids linker; L15—15 amino acids linker; PVX CP—Potato virus X coat protein
The fusions of E6GT with the 4 amino acids linker L4 (amino acids GPGP) (Massa et al. 2008) either at the 3′-terminus of E6GT or at the 5′-terminus of E6GT were prepared by PCR from vector pBSC-E6GT using the primer pairs G9-R and G9-V or G9-Q and G9-O, respectively (Table 1). The first PCR product was cloned into the PVX CP-BH/pMPM-A4Ω plasmid using the XbaI and BglII restriction sites to create the final expression plasmid, which was termed E6GT-L4-PVX CP/pMPM-A4Ω. The product of the second PCR reaction was ligated into the PVX CP-NB/pMPM-A4Ω plasmid using the BglII and SalI restriction sites to produce the final PVX CP-L4-E6GT/pMPM-A4Ω expression plasmid.
The ‘splicing by overlap extension by PCR’ method (Warens et al. 1997) was used to fuse the E6GT coding sequence with the 15 amino acids beta-sheet linker L15 [amino acids (SGGGG)3] (Alfthan et al. 1995) either at the 3′- or at the 5′-terminus. The E6GT sequences (named E6GT-RS and E6GT-NO) were PCR amplified from vector pBSC-E6GT using the primer pairs G9-R and G9-S or G9-N and G9-O, respectively. The L15 linkers (named L15-TJ and L15-AM, respectively) were PCR amplified from vector pTZ-CP-Linker-E7 K + rc (unpublished results) using the primer pairs G9-T and G9-J or G9-A and G9-M, respectively. Two PCR products (either E6GT-RS and L15-TJ or E6GT-NO and L15-AM) were mixed in equimolar amounts. The second-stage PCR was performed using external primers (either G9-R and G9-J or G9-A and G9-M) to produce E6GT-L15 or L15-E6GT products. The E6GT-L15 product was inserted into the PVX CP-BH/pMPM-A4Ω plasmid using the XbaI and BglII restriction sites to create the final expression plasmid, which was termed E6GT-L15-PVX CP/pMPM-A4Ω. The L15-E6GT product was cloned into the PVX CP-NB/pMPM-A4Ω plasmid using the BglII and SalI restriction sites to produce the final PVX CP-L15-E6GT/pMPM-A4Ω expression plasmid (Fig. 1). All PCR reactions were performed using Phusion High-Fidelity DNA polymerase (Finnzymes, Espoo, Finland).
The 3′-terminal part of constructs having the E6GT fused to the 3′-terminus of PVX CP was then inserted into the infectious PVX full length clone pGR106 (kindly provided by DC Baulcombe, The Sainsbury Laboratory, Norwich, UK; Chapman et al. 1992), cleaved by XhoI to produce the PVX CP-E6GT/pGR106, PVX CP-L4-E6GT/pGR106 and PVX CP-L15-E6GT/pGR106 expression plasmids. Thus, these constructs contained the E6GT sequence followed by the repetition of the 3′-terminus of PVX CP and usual pGR106 sequence with its cis acting elements in the 3′-untranslated region. All constructs with E6GT fused to the 5′-terminus of PVX CP were then inserted into pGR106 cleaved by XbaI and XhoI, so the PVX CP present in pGR106 was replaced by chimeric constructs to form E6GT-PVX CP/pGR106, E6GT-L4-PVX CP/pGR106 and E6GT-L15-PVX CP/pGR106, respectively. The constructs were checked for their accuracy by DNA sequencing.
Protein expression in E. coli
The chimeric proteins were expressed from the pMPM-A4Ω plasmid in E. coli MC1061 cells. The optimal conditions for protein expression required incubation for 12 h at 25 °C following induction with 0.2 % arabinose.
Partial purification of PVX CP fused to E6GT – enriched fractions
Fusion proteins between PVX CP and E6GT were partially purified from 500 mL of the bacterial culture. The cells were centrifuged at 15,000×g for 15 min at 4 °C. The pellet was resuspended in 50 mL 20 mM Tris–HCl pH 7.5 with 0.2 mg/mL lysozyme (Sigma) and 0.1 mg/mL DNAse I (Roche), and incubated at 37 °C for 10 min. After homogenisation in glass homogenisator on ice the bacterial lysate was centrifuged. The supernatant containing most of the soluble protein was subjected to further purification on sucrose cushion (Cerovska et al. 2004). The presence of fusion proteins was tested by ELISA (dilution 1 and 10 μg/mL in 15 mM Na2CO3, 35 mM NaHCO3, 3 mM NaN3, pH 9.6). The insoluble fusion proteins were solubilized using 8 M urea. Protein concentration in each fraction was determined by the Bradford method using BSA as a protein standard (Bradford 1976). The fusion proteins from plant material were purified according to Cerovska et al. (2004).
Plant propagation
Four genotypes of Nicotiana benthamiana were evaluated as hosts for expression of modified PVX CPs: N. benthamiana wild type (wt), N. benthamiana carrying the coding sequence of Potato virus A HC-Pro protein (HC-Pro; kindly provided by E. Savenkov, Department of Plant Biology, Sveriges Lantbruksuniversitet, Uppsala, Sweden), N. benthamiana 3H carrying the coding sequence of Tobacco mosaic virus movement protein (3H; kindly provided by R. Beachy, Donald Danforth Plant Science Center, St. Louis, USA) and N. benthamiana K7 plants, which were obtained in our laboratory by crossing N. benthamiana HC-Pro and N. benthamiana 3H. All plants were grown in a growth chamber under controlled conditions (20–25 °C over day, 15–20 °C over night, 16 h-light/8 h-dark). Eighteen plants per group were inoculated in their four-leaf stage. All experiments were performed in triplicate.
Agrobacterium transformation and plant inoculation
The binary vectors with full-length infectious cDNA carrying the PVX CP fusion genes were isolated from E. coli and introduced into chemically competent Agrobacterium tumefaciens (GV3101) using freeze–thaw method (An 1987). Bacteria containing the plasmids were selected on LB-agar plates with 30 μg/mL kanamycin. For transient expression, the resulting Agrobacterium strains induced by acetosyringone were used to infiltrate N. benthamiana leaves. Bacterial suspension for agroinfection of plants was prepared in LB-broth containing 30 μg/mL kanamycin at 28 °C. The primary infection of the host plants was performed by infiltration using 1 mL hypodermic syringe without needle (Hoffmeisterova et al. 2008). As a negative control, leaves inoculated with LB medium were used. As a positive control, N. benthamiana K7 agroinfiltrated with A. tumefaciens containing the pGR106 vector was used.
Polyacrylamide gel electrophoresis (SDS-PAGE)
1 mL aliquots of E. coli MC1061 cells were pelleted by 1 min centrifugation, resuspended in 100 μL of the Laemmli buffer, boiled for 2 min and aliquots were loaded on 12 % polyacrylamide gel containing SDS (Laemmli 1970). The plant samples were homogenized using the FastPrep (MP Biomedicals, Solon, OH USA) system and crude transgenic plant extracts were diluted to a concentration of 1 mg/mL total soluble proteins in the Laemmli buffer containing 6 M urea, placed for 10 min into boiling water, cooled to room temperature and then subjected to SDS-PAGE.
Western blot analysis
The proteins separated by SDS-PAGE were electroblotted to a nitrocellulose membrane (0.2 μm, Sleicher&Schuell Protran) in semidry system (OMNI-TRANS apparatus, Omnibio Brno, Czech Republic) (Hirano and Watanabe 1990). The membrane was then incubated for 1 h in 4 % BSA in PBS + 0.5 % Tween 20 (PBST) and then washed 4 × 5 min in PBST. The primary rabbit polyclonal anti-PVX CP antibodies (dilution 1:1,000 in PBST; PrimeDiagnostics, Wageningen, Netherland) and secondary goat anti-rabbit antibodies conjugated to alkaline phosphatase (AP) (1:30,000 in PBST; Sigma, St. Louis, MO, USA) were used to detect fusion proteins expressed both in E. coli and in N. benthamiana. The primary goat polyclonal anti-HPV16 E6 (N-17) (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the secondary rabbit anti-goat-AP antibodies (1:30,000 in PBST; Sigma, St. Louis, MO, USA) were applied to detect fusion proteins expressed in N. benthamiana. The bands of interest were visualized by reaction with a substrate of AP (BCIP/NBT; Sigma, St. Louis, MO, USA) (Sambrook et al. 1989).
Plate-trapped antigen ELISA (PTA-ELISA)
The PTA-ELISA test was performed as described by Mowat (1985). As primary antibodies the rabbit polyclonal anti-PVX CP (1:1,000; Sigma, St. Louis, MO, USA) or goat anti-HPV16 E6 (N-17) (1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used. As secondary antibodies the AP-conjugated goat anti-rabbit or rabbit anti-goat IgG (Sigma, St. Louis, MO, USA) in dilutions recommended by manufacturers were used. All ELISA measurements were performed in duplicates and repeated three times.
Electron microscopy
Copper grids with carbon support layer were floated on droplets of purified chimeric VLPs (obtained from bacterial or plant expression; 0.8 mg/mL) at room temperature for 40 min. The grids were washed with PBS for 2 min, then with H2O for 2 min (repeated three times). The non-labeled samples were stained with drops of 2 % uranyl acetate at room temperature for 15 min. For the purposes of immunogold-labeling, polyclonal antibodies recognizing PVX CP or E6 were bound to the appropriate parts of the samples by incubating the grid with the rabbit anti-PVX CP IgG (dilution 1:1,000 in PBS; PrimeDiagnostics, Wageningen, Netherland) or goat-anti HPV16 E6 (N-17) (dilution 1:1,000 in PBS; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at room temperature for 40 min. After washing with PBS, samples were decorated with gold-labeled goat anti-rabbit IgG or rabbit—anti goat (10 nm or 15 nm gold particles, Sigma, St. Louis, MO, USA) by floating the grids on a 1:20 PBS dilution of the IgG at room temperature for 40 min. After washing with PBS, the samples were stained with drops of 2 % uranyl acetate (15 min at room temperature). Electron microscopy was carried out using a JEM 1010 transmission electron microscope (Jeol, Japan, facility of Biology Centre of the Academy of Sciences of the Czech Republic, Ceske Budejovice, Czech Republic).
Results
Cloning of fusion constructs
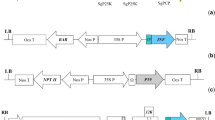
Five different PVX CP constructs (E6GT-PVX CP, E6GT-L4-PVX CP, PVX CP-E6GT, PVX CP-L4-E6GT and PVX CP-L15-E6GT) with the HPV-16 E6GT gene fused either to 5′- or 3′-terminus of PVX CP were prepared (Fig. 1) and then in pGR106 (Fig. 2a, b). We did not obtain the E6GT-L15-PVX CP construct, since in all repeated cloning experiments the L15 linker was about from 21 to 30 nucleotides shorter than expected.

Cloning strategy. a Cloning of 3′-terminally fused constructs into pGR106. To fuse PVX CP with E6GT and linkers L4 or L15 the restriction sites XhoI were used. The final constructs contain the duplicated 3′-terminus of PVX CP. b Cloning of 5′-terminally fused constructs into pGR106. The constructs were cut out from pMPM-A4Ω by XbaI and XhoI and inserted into pGR106 cleaved by NheI and XhoI. LB left border, RB right border, 35S Cauliflower mosaic virus 35S promoter, RdRp RNA-dependent RNA polymerase, 25 K, 12 K and 8 K triple gene block proteins, PVX CP Potato virus X coat protein, CP promoter promoter of PVX CP, Nos nopaline synthase terminator, E6GT mutated form of E6 from HPV16, L4 s4 amino acids linker; L15 15 amino acids linker
Expression and purification of fusion proteins in E. coli
All fused proteins were expressed at high levels in E. coli except for the E6GT-L15-PVX CP. The theoretical molecular weight of the fusion proteins was approximately 44 kDa, with small differences caused by the presence of linkers, in the case of L4 about 0.3 kDa and in the case of L15 about 1 kDa. All fusion proteins expressed in E. coli showed bands of the appropriate size on the Western blots (Fig. 3a, b).

Western blot analysis of fusion proteins PVX CP E6GT expressed in bacteria and plants. a Western blot analysis of N-terminally fused proteins PVX CP E6GT expressed in E. coli MC1061. 1 pMPM-A4Ω in MC1061; 2 positive control (E7GGG-PVX CP); M M Spectra™ Multicolor Broad Range Protein Ladder (10–260 kDa), Fermentas; 3 E6GT-PVX CP before induction; 4 E6GT-L4-PVX CP before induction; 5 E6GT-PVX CP after induction; 6 E6GT-L4-PVX CP after induction. b Western blot analysis of C-terminally fused proteins PVX CP E6GT expressed in E. coli MC1061. M M Spectra™ Multicolor Broad Range Protein Ladder (10–260 kDa), Fermentas; 1 PVX CP in MC1061; 2 positive control (E7GGG-PVX CP); 3 PVX CP-L15-E6GT before induction; 4 PVX CP-L15-E6GT not induced; 5 PVX CP-L15-E6GT after induction; 6 PVX CP-E6GT before induction; 7 PVX CP-E6GT without induction; 8 PVX CP-E6GT after induction; 9 PVX CP-L4-E6GT before induction; 10 PVX CP-L4-E6GT without induction; 11 PVX CP-L4-E6GT after induction. c Western blot analysis of C-terminally fused proteins PVX CP E6GT expressed in N. benthamiana K7 plants. 1 250 ng of purified PVX; 2 250 ng of PVX CP-L4-E6GT purified from E. coli; 3 not infected plant; M M Spectra™ Multicolor Broad Range Protein Ladder (10–260 kDa), Fermentas; 4 PVX CP-L15-E6GT in inoculated leaves 12 days after inoculation (dpi); 5 PVX CP-E6GT in inoculated leaves 12 dpi; 6 PVX CP-L4-E6GT in inoculated leaves 12 dpi
The yields of purified C-fusion proteins expressed in E. coli were 1.85 ± 0.4 mg/mL. In comparison with the C-fusions significantly larger quantities of N-fusion proteins were produced in E. coli inclusion bodies. The yields of the N-terminal fusion proteins (both the soluble part and pellets) were 2.3 ± 0.4 mg/mL (P < 0.05).
Although most of the N-terminal fusion proteins were soluble, a small part of them remained in the pellet; purification of C-terminal fusion proteins showed higher amounts of recombinant proteins in the soluble fraction.
Production of virus-like particles in E. coli
The partially purified fusion proteins expressed in E. coli were subjected to electron microscopy. The chimeric VLPs were visualized with 2 % uranyl acetate. The putative VLPs showed similar shape and length as the other particles formed by recombinant PVX CP expressed in E. coli (Maclean et al. 2007; Fernández-San Millán et al. 2008). Immunogold-labeling using polyclonal antibodies against PVX CP proved that all our fusion proteins form chimeric particles. For illustration, chimeric VLPs of expressed fusion protein PVX CP-L4-E6GT are shown in Fig. 4a.

Immunoelectron microscopy of chimeric virus-like particles of expressed fusion protein PVX CP-L4-E6GT using negative staining by 2 % uranyl acetate. a Immunochemical labeling of PVX CP-L4-E6GT isolated from E. coli MC1061. The particles were decorated with rabbit anti-PVX CP (1:1,000 in PBS; PrimeDiagnostics, Wageningen, Netherland); secondary goat anti-rabbit 10 nm gold conjugate (1:20 in PBS; Sigma, St. Louis, MO, USA). Bar represents 500 nm; b Immunochemical labeling of PVX CP-L4-E6GT partially purified from N. benthamiana K7 plants. The particles were decorated with polyclonal goat anti-HPV16 E6 (N-17) antibodies (1:1,000 in PBS; Santa Cruz Biotechnology, Santa Cruz, CA, USA), secondary rabbit anti-goat antibodies conjugated with 10 nm Au (1:20; Sigma, St. Louis, MO, USA), polyclonal rabbit anti-PVX CP antibodies (1:1,000 in PBS; PrimeDiagnostics, Wageningen, Netherland), secondary goat anti-rabbit antibodies conjugated with 5 nm Au (1:20 in PBS; Sigma, St. Louis, MO, USA). Bar represents 200 nm
Selection of the most suitable plant genotype
The N. benthamiana K7 plants were shown to provide the highest expression of target proteins, about 50-times more than wt and HC-Pro plants and about twice as much as 3H plants, as evidenced by SDS-PAGE/Western blot analysis. For illustration, expression of fusion protein PVX CP-L15-E6GT is shown in Fig. 5.

Western blot analysis of fusion protein PVX CP-L15-E6GT expressed in N. benthamiana plants. M M Spectra™ Multicolor Broad Range Protein Ladder (10–260 kDa), Fermentas; 1 N. b. inoculated leaves; 2 N. b. systemic leaves; 3 N. b. roots; 4 N. b. HC-Pro inoculated leaves; 5 N. b. HC-Pro systemic leaves; 6 N. b. HC-Pro roots; 7 N. b. 3H inoculated leaves; 8 N. b. 3H systemic leaves; 9 N. b. 3H roots; 10 N. b. K7 inoculated leaves; 11 N. b. K7 systemic leaves; 12 N. b. K7 roots. To detect the fusion protein the primary rabbit polyclonal anti-PVX CP antibodies were used
Expression and purification of fusion proteins in N. benthamiana K7 plants
All constructs were subsequently expressed in N. benthamiana K7 plants. The infection was initiated by agroinfiltration using a syringe without needle (Hoffmeisterova et al. 2008). In the following mechanical inoculations the extracts from agroinfiltrated leaves were not infectious.
The presence of fusion genes and their products in harvested N. benthamiana K7 leaves was determined by immunocapture (IC) RT-PCR (Fig. 6), plate-trapped antigen (PTA) ELISA and Western blot, using IgG antibodies specific to PVX CP and E6. The ELISA absorbances of samples prepared from leaves inoculated with LB medium (negative controls) were comparable to the absorbances of healthy leaves without LB treatment.

Immunocapture RT-PCR analysis of fusion genes in inoculated N. benthamiana K7 leaves. M GeneRuler 1 kb DNA Ladder, Fermentas; 1 PVX CP-L15-E6GT; 2 PVX CP-E6GT; 3 PVX CP-L4-E6GT; 4 PVX CP; 5 healthy leaves; 6 E6GT-PVX CP; 7 E6GT-L4-PVX CP; 8 healthy leaves; 9 PVX CP. The E6GT (G9-O) and pGR106-specific primers were used to amplify 3′-terminal fusion genes (1–5). The pGR106-specific primers were used to detect 5′-terminal fusion genes (6–9)
We could not confirm the expression of N-terminal fusion proteins in plants by immunoblotting, therefore we isolated total RNA from the appropriate infected leaves and performed RT-PCR. After sequencing the RT-PCR products, we found out that nucleotide sequences of the E6GT fused to the 5′-terminus of PVX CP were mutated. The mutations occured in the region of nucleotides 170–324 in the case of the E6GT-PVX CP construct and in the region of nucleotides 74–324 in the case of the E6GT-L4-PVX CP construct. Both regions accumulated different nucleotide substitutions that resulted in amino acid changes and premature stop codons. It seems that after the infection of plants the PVX virus does not tolerate the 5′-terminal E6GT fusion.
On the contrary, the presence of all C-terminal fusion proteins in N. benthamiana K7 plants was confirmed both by PTA ELISA and Western blot analysis with antibodies against PVX CP and E6 (Fig. 3c) and by IC RT-PCR (Fig. 6). The amounts of chimeric proteins were determined by SDS-PAGE/Western blot analysis using anti-E6 antibody (Fig. 7). The levels of all expressed C-terminal fusion proteins were determined using serial dilutions of PVX CP-L4-E6GT expressed in bacteria as a standard. The average yields of all C-terminal PVX CP fusion proteins after purification were approximately 5 ± 1.2 mg/kg (P < 0.05) of fresh leaf tissue.

Transient expression of three PVX CP C-terminus fusion proteins in Nicotiana benthamiana K7 plants. N. benthamiana K7 leaves were agroinfiltrated and the accumulation of each fusion protein was quantified by Western blot analysis using anti-E6 (N-17) antibodies (1:1,000 in PBST; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and by rabbit anti-goat-AP secondary antibodies (1:30,000 in PBST; Sigma, St. Louis, MO, USA), where the bands corresponding to the respective constructs were compared with the dilution series of the PVX CP-L4-E6GT expressed in bacteria. Each column represents the mean value of 3 independent experiments. In each experiment 2 agroinfiltrated leaves per plant (18 plants in total) were collected. The standard deviation is represented by error bars. TSP total soluble protein
Western blot analysis of different plant parts showed that the fusion proteins were present only in inoculated leaves and that the infection did not spread systemically as did the wild type virus (Fig. 5).
Production of virus-like particles in plants
Immunogold-labeling with polyclonal antibodies against PVX CP and E6 confirmed the presence and accessibility of the desired epitopes on the surface of the purified particles. Chimeric VLPs of PVX CP-L4-E6GT fusion protein are shown for illustration (Fig. 4b).
Discussion
Cervical carcinomas are almost universally associated with high-risk human papillomavirus infections and are a leading cause of cancer death in women worldwide. HPV oncoproteins contribute to cancer initiation and progression and their expression is necessary for the maintenance of the transformed state. Disease prevention has been realized by introduction of prophylactic vaccines that prevent transmission of specific high-risk HPVs. However, these vaccines have no therapeutic efficacy and since HPV-associated cervical cancers may arise years or even decades after the initial infection, it has been estimated that there will be no measurable decline of HPV-associated tumors before 2040. Hence, therapeutic efforts to combat high-risk HPV-associated disease remain of critical importance (Hellner and Munger 2011).
High-risk human papillomavirus infections are generally transient and believed to be controlled by cell-mediated immune response because the majority of HPV-induced lesions spontaneously regress (Eiben et al. 2003). Nevertheless, a major feature of HPV that might impair HPV therapeutic vaccination is the poor presentation of viral antigens, which are expressed at a low level (Tindle 2002). As the virus cannot be propagated in culture, vaccines have been based on recombinant antigens with inherent high-cost production. Therefore, in a search for alternative formulations able to improve the immunogenicity of HPV-16 E6 protein, we took advantage of our extensive experience with PVX based expression platform (Cerovska et al. 2004, 2008, 2012; Pokorná et al. 2005; Hoffmeisterova et al. 2008; Plchova et al. 2011) and used this system for the transient expression of chimeric PVX particles bearing mutagenized HPV-16 E6 in plants. Other studies have also demonstrated the advantage of PVX as an epitope presentation system in vaccination (Franconi et al. 2002; Marusic et al. 2001).
We successfully expressed all full-length C- and two N-terminal (no linker, L4) fusion proteins in E. coli MC1061 using the pMPM-A4Ω plasmid.
After optimization of expression conditions in E. coli high yield and proper serological reactivity of all constructs were observed. Both N-terminal and C-terminal fusion proteins accumulated to approximately the same levels. Also the length of the linker did not have substantial impact on yield of fusion proteins. Based on immunoelectron microscopy analysis with anti-PVX CP antibodies we can conclude that all fusion proteins were able to form VLPs.
On the contrary, only the C-terminal fusion proteins accumulated to detectable levels in plants. The highest yields of chimeric protein were observed in the transgenic N. benthamiana K7. When inoculated on host plants, these recombinant viruses were not able to spread systemically, the viral infection was restricted only to the inoculated leaves. All three constructs were expressed in plant tissue at approximately similar levels and there were no substantial differences when linkers of different lengths were used. All three fusion proteins expressed in plants formed VLPs with serologically accessible parts of PVX CP and E6GT protein.
The main advantage of transient-expression system in plants is the relative rapidity of the procedure, while the main constraint of this approach is generally a low stability and infectivity of the chimeric structures carrying exogenous sequences fused to the N-terminus of PVX CP (Uhde-Holzem et al. 2007). We have previously expressed E7GGG fused to both N- and C-terminus of PVX CP in N. benthamiana HC-Pro transgenic plants (Plchova et al. 2011). The chimeric coat proteins carrying E7GGG fusions were expressed in plant tissues at approximately the same concentrations and, similarly to E6 fusions, were not able to cause infection of upper systemic leaves. This observation correlated with inability of E7GGG to form viral particles probably due to the steric obstructions or change in the charge.
On the contrary, in this work, the attenuated E6GT HPV oncoprotein accumulated to detectable levels only when fused to the C-terminus of PVX CP. Again, the fusion proteins were not able to spread systemically in transgenic K7 N. benthamiana, even though all of them were able to form viral particles as evidenced by electron micrographs and IC-RT-PCR. The K7 plants carry both HC-Pro from PVA and MP from TMV. HC-Pro, which is known as a viral suppressor of RNA silencing is known to elevate the level of potexvirus expression while the TMV MP is known to complement the lack of cell-to-cell transport activity of wild type PVX CP (Fedorkin et al. 2001). Both these traits have likely contributed to obtain the highest expression in this host genotype.
We could only speculate why the chimeric proteins, that formed the particles, were unable to spread systemically. It is possible that the characteristics of the fusion with E6GT (e.g. fused sequence length and amino acid composition), and/or its location in the vector prevented the virus from interacting with some host factors, which are indispensable for long-distance movement or entry from phloem to cells.
In our future research we would like to address the instability of N- and C-terminal PVX-CP fusions. One of the possible strategies we would like to follow is to identify novel positions in the PVX-CP sequence allowing stable surface presentation of heterologous antigens. Also the comparison of immunogenicity of VLPs with that of peptide vaccines containing corresponding E6-derived epitopes is of great interest. Last but not least combined immunization with DNA vaccines and VLPs will also be tested, as this heterologous prime/boost strategy proved to be very efficient in other systems (Woodland 2004).
References
Alfthan K, Takkinen K, Sizmann D, Soderlund H, Teeri TT (1995) Properties of a single-chain antibody containing different linker peptides. Protein Eng 8:725–731
An G (1987) Binary Ti vectors for plant transformation and promoter analysis. Methods Enzymol 153:292–305
Anwar N, Watanabe KN, Watanabe JA (2011) Transgenic sweet potato expressing mammalian cytochrome P450. Plant Cell Tiss Organ Cult 105:219–231
Biemelt S, Sonnewald U, Galmbacher P, Willmitzer L, Muller M (2003) Production of human papillomavirus type 16 virus-like particles in transgenic plants. J Virol 77:9211–9220
Billich A (2003) HPV vaccine MedImmune/GlaxoSmithKline. Curr Opin Investig Drugs 4:210–213
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Canizares MC, Nicholson L, Lomonossoff GP (2005) Use of viral vectors for vaccine production in plants. Immunol Cell Biol 83:263–270
Cerovska N, Pecenkova T, Moravec T, Veleminsky J (2004) Transient expression of heterologous model gene in plants using Potato virus X-based vector. Plant Cell Tissue Organ Cult 79:147–152
Cerovska N, Hoffmeisterova H, Pecenkova T, Moravec T, Synkova H, Plchova H, Ludvikova V, Smahel MK (2008) Transient expression of HPV16 E7 peptide (aa 44–60) and HPV16 L2 peptide (aa 108–120) on chimeric potyvirus-like particles using Potato virus X-based vector. Protein Expr Purif 58:154–161
Cerovska N, Hoffmeisterova H, Moravec T, Plchova H, Folwarczna J, Synkova H, Ryslava H, Ludvikova V, Smahel M (2012) Transient expression of Human papillomavirus type 16 L2 epitope fused to N- and C-terminus of coat protein of Potato virus X in plants. J Biosci 37:125–133
Chapman S, Kavanagh TA, Baulcombe DC (1992) Potato virus X as a vector for gene expression in plants. Plant J 2:549–557
De la Rosa GP, Monroy-García A, de Lourdes Mora-García M, Reynaga Peńa CG, Hernández-Montes J, Weiss-Steider B, Gómez Lim MA (2009) An HPV 16 L1-based chimeric human papilloma virus-like particles containing a string of epitopes produced in plants is able to elicit humoral and cytotoxic T-cell activity in mice. Virol J 6:2. doi:10.1186/1743-422X-6-2
Eiben GL, Da Silva DM, Steven DM, Fausch C, Le Poole IC, Nishimura MI, Kast WM (2003) Cervical cancer vaccines: recent advances in HPV research. Viral Immunol 16:111–121
Faye L, Gomord V (2010) Success stories in molecular farming—a brief overview. Plant Biotechnol J 8:525–528
Fedorkin ON, Solovyev AG, Yelina NE, Zamyatin AA, Inovkin RA, Makinen K, Schieman J, Syu M (2001) Cell-to-cell movement of Potato virus X involves distinct functions of the coat protein. J Gen Virol 82:449–458
Fernández-San Millán A, Ortigosa SM, Hervás-Stubbs S, Corral-Martínez P, Seguí-Simarro JM, Gaétan J, Coursaget P, Veramendi J (2008) Human papillomavirus L1 protein expressed in tobacco chloroplasts self-assembles into virus-like particles that are highly immunogenic. Plant Biotechnol J 6:427–441
Franconi R, DiBonito P, Dibello F, Accardi L, Muller A, Cirilli A (2002) Plant derived human papillomavirus 16 E7 oncoprotein induces immune response and specific tumor protection. Cancer Res 62:3654–3658
Frazer H (2004) Prevention of cervical cancer through papillomavirus vaccination. Nat Rev Immunol 4:46–54
Ganguly N, Parihar SP (2009) Human papillomavirus E6 and E7 oncoproteins as risk factors for tumorigenesis. J Biosci 34:113–123
Harper DM, Franco EL, Wheeler CM, Moscicki AB, Romanowski B, Roteli-Martins CM, Jenkins D, Schuind A, Costa Clemens SA, Dubin G (2006) Sustained efficacy up to 4.5 years of a bivalent L1 virus-like particle vaccine against human papillomavirus types 16 and 18: follow-up from a randomised control trial, HPV Vaccine Study group. Lancet 367:1247–1255
Hellner K, Munger K (2011) Human papillomaviruses as therapeutic targets in human cancer. J Clin Oncol 29:1785–1794
Hirano H, Watanabe T (1990) Microsequencing of proteins electrotransferred onto immobilizing matrices from polyacrylamide gel electrophoresis: application to an insoluble protein. Electrophoresis 11:573–580
Hoffmeisterova H, Cerovska N, Moravec T, Plchova H, Folwarczna J, Veleminsky J (2008) Transient expression of fusion gene coding for the HPV-16 epitopes fused to the sequence of potyvirus coat protein using different means of inoculation of Nicotiana benthamiana and Brassica rapa, cv. Rapa plants. Plant Cell Tissue Organ Cult 94:261–267
Hoffmeisterova H, Moravec T, Plchova H, Folwarczna J, Cerovska N (2012) The influence of the N- and C-terminal modifications of Potato virus X coat protein on virus properties. Biol Plant 56:775–779
Hsu CH, Peng KL, Jhang HC, Lin CH, Wu SY, Chiang CM, Lee SC, Yu WC, Juan LJ (2011) The HPV E6 oncoprotein targets histone methyltransferases for modulating specific gene transcription. Oncogene 31:2335–2349
Kim MY, Kim TG, Yoo HS, Yang MS (2011) Expression and assembly of ApxIIA toxin of Actinobacillus pleuropneumoniae fused with the enterotoxigenic E. coli heat-labile toxin B subunit in transgenic tobacco. Plant Cell Tissue Organ Cult 105:375–382
Koutsky LA, Ault KA, Wheeler CM, Brown DR, Bar E, Alvarez FB, Chiacchierini LM, Jansen KU (2002) A controlled trial of a human papillomavirus type 16 vaccine. Proof of principle study investigators. N Engl J Med 347:1645–1651
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Lico C, Capuano F, Renzone G, Donini M, Marusic C, Scaloni A, Benvenuto E, Baschieri S (2006) Peptide display on Potato virus X: molecular features of the coat protein- fused peptide affecting cell-to-cell and phloem movement of chimeric virus particles. J Gen Virol 87:3103–3112
Lico C, Mancinia C, Italiani P, Betti C, Boraschib D, Benvenuto E, Baschieri S (2009) Plant-produced Potato virus X chimeric particles displaying an influenza virus-derived peptide activate specific CD8+ T cells in mice. Vaccine 27:5069–5076
Liu HL, Li WS, Lei T, Zheng J, Zhang Z, Yan XF, Wang ZZ, Wang IL, Si LS (2005) Expression of human papillomavirus type 16 L1 protein in transgenic tobacco plants. Acta Biochim Biophys Sin 37:153–158
Ma B, Xu Y, Hung C-F, Wu T-C (2010) HPV and therapeutic vaccines: where are we in 2010? Curr Cancer Ther Rev 6:81–103
Maclean J, Koekemoer M, Olivier AJ, Stewart D, Hitzeroth II, Rademacher T, Fischer R, Williamson AL, Rybicki EP (2007) Optimization of human papillomavirus type 16 (HPV-16) L1 expression in plants: comparison of the suitability of different HPV-16 L1 gene variants and different cell-compartment localization. J Gen Virol 88:1460–1469
Marconi G, Albertini E, Barone P, De Marchis F, Lico C, Marusic C, Rutili D, Veronesi F, Porceddu A (2009) In planta production of two peptides of the Classical Swine Fever Virus (CSFV) E2 glycoprotein fused to the coat protein of Potato virus X. BMC Biotechnol 6:1–9
Martínez-González L, Rosales-Mendoza S, Soria-Guerra RE, Moreno-Fierros L, López-Revilla R, Korban SS, Guevara-Arauza JC, Alpuche-Solís AG (2011) Oral immunization with a lettuce-derived Escherichia coli heat-labile toxin B subunit induces neutralizing antibodies in mice. Plant Cell Tiss Organ Cult 107:441–449
Marusic C, Rizza P, Lattanzi L, Mancini C, Spada M, Belardelli F, Benvenuto E, Capone I (2001) Chimeric plant virus particles as immunogens for inducing murine and human immune responses against human immunodeficiency virus type 1. J Virol 75:8434–8439
Massa S, Simeone P, Muller A, Benvenuto E, Venuti A, Franconi R (2008) Antitumor activity of DNA vaccines based on the human papillomavirus-16 E7 protein genetically fused to a plant virus coat protein. Hum Gene Ther 19:354–364
McLaughlin-Drubin ME, Munger K (2009) Oncogenic activities of human papillomaviruses. Virus Res 143:195–208
Morgenfeld M, Segretin ME, Wirth S, Lentz E, Zelada A, Mentaberry A, Glossmann L, Almonacid FB (2009) Potato virus X coat protein fusion to human papillomavirus 16 E7 oncoprotein enhance antigen stability and accumulation in tobacco chloroplast. Mol Biotechnol 43:243–249
Mowat WP (1985) Simplified enzyme immunoassay for plant virus detection and identification. In: Harrison BD, Murant AF (eds) Report of the Scottish crop Researche Institute for 1984. Scottish Crop Research Institute, Dundee, p 188
Münger K, Phelps WC, Bubb V, Howley PM, Schlegel R (1989) The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primer human keratinocytes. J Virol 63:4417–4421
Plchova H, Moravec T, Hoffmeisterova H, Folwarczna J, Cerovska N (2011) Expression of Human papillomavirus 16 E7ggg oncoprotein on N- and C-terminus of Potato virus X coat protein in bacterial and plant cells. Protein Expr Purif 77:146–152
Pokorná D, Čeřovská N, Šmahel M, Moravec T, Ludvíková V, Macková J, Synková H, Dušková M, Hozák P, Velemínský J (2005) DNA vaccines based on chimeric potyvirus-like particles carrying HPV16 E7 peptide (aa 44–60). Oncol Rep 14:1045–1053
Polakova I, Pokorna D, Duskova M, Smahel M (2010) DNA vaccine against human papillomavirus type 16: modifications of the E6 oncogene. Vaccine 28:1506–1513
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Scholthof K-BG, Mirkov TE, Scholthof HB (2002) Plant viral gene vectors: biotechnology applications in agriculture and medicine. Genet Eng 24:67–85
Tindle RW (2002) Immune evasion in human papillomavirus-associated cervical cancer. Nature Rev Cancer 2:59–65
Uhde-Holzem K, Fischer R, Commandeur U (2007) Genetic stability of recombinant potato virus X virus vectors presenting foreign epitopes. Arch Virol 152:805–811
Varsani A, Williamson AL, Rose RC, Jaffer M, Rybicki EP (2003) Expression of Human papillomavirus type 16 major capsid protein in transgenic Nicotiana tabacum cv, Xanthi. Arch Virol 148:1771–1786
Villa LL, Costa RL, Petta CA, Andrade RP, Ault KA, Giuliano AR, Wheeler CM, Koutsky LA, Malm C, Lehtinen M, Skjeldestad FE, Olsson SE, Steinwall M, Brown DR, Kurman RJ, Ronnett BM, Stoler MH, Ferenczy A, Harper DM, Tamms GM, Yu J, Lupinacci L, Railkar R, Taddeo FJ, Jansen KU, Esser MT, Sings HL, Saah AJ, Barr E (2005) Prophylactic quadrivalent human papillomavirus (types 6, 11, 16, and 18) L1 virus-like particle vaccine in young women: a randomised double-blind placebo-controlled multicentre phase II efficacy trial. Lancet Oncol 6:271–278
Warens AN, Jones MD, Lechler RI (1997) Splicing by overlap extension by PCR using asymmetric amplification improved technique for the generation of hybrid proteins of immunological interest. Gene 186:29–35
Woodland DL (2004) Jump-starting the immune system: prime-boosting comes of age. Trends Immunol 25:98–103
Acknowledgments
This research was supported by the grants No. 521/09/1525 and No. P501/12/1761 of the Grant Agency of the Czech Republic. We acknowledge the skilled technical assistance of Mrs. R. Hadamkova and D. Cibochova.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cerovska, N., Moravec, T., Hoffmeisterova, H. et al. Expression of a recombinant Human papillomavirus 16 E6GT oncoprotein fused to N- and C-termini of Potato virus X coat protein in Nicotiana benthamiana . Plant Cell Tiss Organ Cult 113, 81–90 (2013). https://doi.org/10.1007/s11240-012-0253-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-012-0253-3