
Abstract
Nickel(II) complex with sarcosine [Ni(sar)2(H2O)2] (1) has been obtained as a reaction product of a system: Ni(II)Cl2–1-methylhydantoin. Basic hydrolysis of the organic substrate leads to in situ formation of sarcosine ligand, which coordinates to the metal ion. The isolated complex has been characterized by means of single crystal X-ray diffraction and spectroscopic studies (IR, Raman, and NIR–UV–Vis) supported with DFT calculations. Single crystal X-ray diffraction revealed that the nickel environment in [NiN2O4] chromophore exhibited the geometry of a tetragonally elongated octahedron. Hydrogen bonds create two chain patterns which propagate along the main crystallographic directions a and b. It leads to the formation of a 2D network. Electronic spectra analysis showed that the nickel surroundings can be described as pseudooctahedral in solution and tetragonal in the solid state. Based on the calculated 10Dq parameter (10,990 cm−1), sarcosine ligand was located in the spectrochemical series close to ammonia. Comprehensive studies of the molecular structure and vibrational spectra of the title complex have been performed using UPBE0, unrestricted density functional method. The clear-cut assignment of the bands in FT-IR and Raman spectra of studied complex has been made on the basis of the calculated potential energy distribution. The Ni–L(sarcosine) stretching vibrations were assigned in Raman spectrum to the medium intensity band at 442 cm−1.
Similar content being viewed by others
Introduction
Sarcosine (N-methylglycine) (Fig. 1) is an α-amino acid occurring in various living organisms as an intermediate in amino acid metabolism [1] and as a component of peptides, e.g., actinomycines [2]. Additionally, it can potentially serve as a liposome cryoprotectant and as a drug in the treatment for schizophrenia [3, 4].

Scheme of the hydrolysis of 1-methylhydantoin, which results in a sarcosine as the final products
In recent years, various sarcosine adducts, salts, and metal complexes have been prepared. The latter have been studied in order to understand the mechanism of interactions between amino acids and metal ions found in biological systems, i.e., Ca, Zn, Cu [5–7]. They can serve as potential laser materials [8] as well as anticancer drugs [9]. In all cases, they have been prepared using sarcosine as a starting reagent.
Sarcosine can also be a product of acid or basic hydrolysis of 1-methylhydantoin (Fig. 1). Hydantoin and its derivatives at moderate temperature and pH form complexes with metal ions [10–13]. However, when elevated temperature and extreme pH are applied, hydantoins hydrolyze, forming in the most cases, amino acids [14]. Such in situ formed amino acids could subsequently coordinate to metal ions [15].
In the present work, the complex [Ni(sar)2(H2O)2] (1) has been obtained as a result of the reaction between 1-methylhydantoin and nickel(II) salt. At elevated temperature and pH, 1-methylhydantoin hydrolyzed resulting in a sarcosine, ammonia, and carbon dioxide as the final products (Fig. 1) [14]. Then, in situ produced ligand was coordinated by metal ion forming (1). Incomplete structure of (1) was known earlier [16]. Here, it has been characterized in detail using both experimental (X-ray diffraction, IR, Raman, and electronic spectroscopy) and theoretical (DFT) methods. To the best of our knowledge, this is the first example of a sarcosine complex synthesized from an organic substrate other than sarcosine itself. It was found that crystals of (1) obtained by this “indirect” method are of better quality than those created from sarcosine as a starting reagent. For example, they do not show tendency to twinning.
Experimental
Preparation
1-Methylhydantoin was purchased from Sigma-Aldrich, while nickel(II) chloride (anhyd.) and ammonia solution (25 %) were from POCH S.A. and used without further purification. Nickel(II) chloride (0.5 mmol) and 1-methylhydantoin (1 mmol) were dissolved in 10 cm3 of ammonia solution and then heated in the solvothermal conditions at 383 K for 15 h. After cooling down, the resultant solution was left to slow evaporation in the air. After 1 month, blue single crystals of (1) were obtained.
Elemental analysis
Elemental analysis was carried out using elemental analyzer Vario El III (Elementar). Anal. Calc. for C6H16N2NiO6 (%): C, 26.60; N, 10.34; H, 5.95. Found: C, 26.60; N, 10.31; H, 5.98.
X-ray diffraction
X-ray diffraction data were collected on a KUMA Diffraction KM-4 four-circle single crystal diffractometer equipped with a CCD detector using graphite-monochromatized MoK α radiation (α = 0.71073 Å). Experiment was carried out at 295 K. The raw data were treated with the CrysAlis Data Reduction Program (version 1.172.33.42) taking into account an absorption correction. The intensities of the reflection were corrected for Lorentz and polarization effects. The crystal structure was solved by direct methods [17] and refined by full-matrix least-squares method using SHELXL-2013 and ShelXle programs [17, 18] (Table 1). Non-hydrogen atoms were refined using anisotropic displacement parameters. H-atoms of the sarcosine anion were visible on the Fourier difference maps, but placed by geometry and allowed to refine “riding on” the parent atom. The positions of H-atoms of the water molecule and NH group were refined without constraints, but Uiso(H) = 1.5Ueq(O) and Uiso(H) = 1.2Ueq(N).
NIR–UV–Vis electronic, IR, and Raman spectroscopy
Electronic spectra of solid state (reflectance) and water solution (absorbance) (c = 6.94 10−3 M) were recorded on Cary 500 Scan (Varian) UV–Vis–NIR spectrophotometer in the range of 7500–35,000 cm−1, with resolution of 10 cm−1. To obtain accurate band positions, the spectra were analyzed using variable digital filter method [19, 20] with the following parameters: a real number determining the degree of resolution enhancement, α = 200; the integer number determining the filter width, N = 10; the increment between points (step) = 100 cm−1. Crystal field parameters were calculated based on the known equations for 3d8 configuration with Oh and D4h symmetry of the metal surrounding [21, 22].
The FT-IR spectrum was measured on Bruker IFS 113 V spectrometer in the region of 4000–400 cm−1 with resolution of 2 cm−1 using KBr pellets. The far-infrared spectrum, in the range of 600–50 cm−1, was recorded on FT-IR Bruker IFS 66/S spectrometer with resolution of 2 cm−1 using Nujol mull technique. The FT-Raman spectrum, in the range of 4000–50 cm−1, was measured on Bruker MultiRAM spectrometer equipped with Nd:YAG laser, emitting radiation at a wavelength of 1064 nm and liquid nitrogen-cooled germanium detector. The spectrum was recorded with resolution of 2 cm−1.
Theoretical study
The molecular structure of (1) has been fully optimized with unrestricted density functional one-parameter hybrid protocol PBE0 [23–25]. The combined basis sets were used in calculation: the polarized valence double-ζ basis set D95v(d,p) for nonmetal atoms and for nickel the LanL2DZ effective core potential with conjunct valence basis set [26]. The examined value of total spin, Ŝ2, was equal to 2.000, which corresponds to a triplet ground-state wave function with no spin contamination [27]. All calculations have been performed using the Gaussian 09 set of programs [28]. Natural charges were obtained by the natural bond orbital (NBO) analysis using version 5.0 of the program [29, 30].
For clear-cut vibrational assignment of the experimental spectra of (1), a normal coordinate analysis was applied. The potential energy distribution (PED) was calculated with the procedure as described earlier [31, 32]. A non-redundant set of 87 internal coordinates has been constructed, as suggested by Fogarasi et al. [33]. PED calculations were performed using the Balga program [34]. The theoretical frequencies above 1510 cm−1 have been scaled.
The theoretical Raman intensities IR were calculated according to the following formula [35–37]:
where C is a constant equal to 10−12, B i is the temperature factor represented by the Boltzmann distribution (in this work assumed to be 1 as discussed earlier [37]), υ 0 is the wave number of the laser radiation (in this work, υ 0 = 9398.5 cm−1, which corresponds to a wavelength of the 1064-nm line of a Nd:YAG laser), υ i is the wave number of the normal mode (cm−1), and Si is the computed Raman scattering activity of normal mode Q i . The calculated Raman intensities I R presented in Table 6 are given in arbitrary units.
Results and discussion
Examination of Cambridge Structural Database (CSD) (version 5.34) revealed that the sarcosine moiety may occur in various forms in complexes: an anion, a zwitterion, or a cation and its various forms coordinate in different ways. When anionic form is present, sarcosine forms a bidentate ligand where N and one of O atoms are involved in coordination (Fig. 2a) [9, 38–40]. In the case of zwitterion and cationic forms, it acts as mono- [7], bidentate [8] or bridging ligand [5, 41–43], but only carboxylate group binds to metal (Fig. 2b, c). Among metal complexes with sarcosine, cationic form of ligand is the least common. Anionic and zwitterion forms are much more abundant, especially as bidentate or bridging ligands.

Forms of sarcosine ligand and types of the coordination in sarcosine metal complexes: a anionic bidentate ligand coordinating by the N and one of the O atoms, b zwitterion mono- or bidentate, sometimes bridging, ligand, c cationic monodentate ligand
Crystal structure
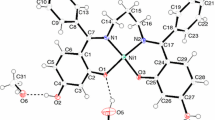
The structural parameters of (1) were published a long time ago, but the positions of the hydrogen atoms were not reported [16]. Therefore, the crystal structure of this compound is presented de novo in this paper. The complex crystallizes in centrosymmetric space group P-1 of the triclinic symmetry (Table 1). Two sarcosine anions bind through amino and carboxylate groups, and with two water molecules create a distorted octahedral arrangement around the nickel ion (Fig. 3). Half of the (1) molecule lies in the asymmetric part of the unit cell because the nickel ion lies on the inversion center and therefore (1) possesses C i point group symmetry. Table 2 shows that Ni–N1 and Ni–O1 bond lengths are significantly shorter than Ni–O1W distances resulting in tetragonal elongation of the nickel(II) octahedron along two Ni–O1W bonds.

a Crystal structure of [Ni(sar)2(H2O)2]. b A view of [Ni(sar)2(H2O)2] molecules arranged in layers parallel to ab plane
In the crystal structure of (1), N–H···O and O–H···O hydrogen bonds join adjacent molecules arranged in a two-dimensional network of hydrogen bonds (Fig. 3b, Table 3). The network results in an intersection of the chain patterns which propagate along the main crystallographic directions, a and b (Fig. 4a, b). Each and every chain contains only one type of hydrogen bond, and therefore, these chain patterns described by the unitary graph-set descriptors are the most important one in the crystal structure of (1) [44]. Additionally, ring patterns are also present, two R 22 (8) and one R 22 (12). Although two rings R 22 (8) are described by the same descriptor, they arise from different summation of the elementary graph-set descriptors because different atomic pathways are related to each pattern [45]. The first one results from summation E 02 (5)HNNiOH + E 20 (3)OCO = R 22 (8) and the second one from 2·E 11 (4)HONiO = R 22 (8) (Fig. 4a). Since two chains C(6) intersect each other at the nickel ion (Fig. 4b), the ring pattern R 22 (12) is created. It is connected to the relation 2·E 11 (6)HONiOCO = R 22 (12). It is worth noting that R 22 (12) ring and the second R 22 (8) ring result from doubling E 11 (6)HONiOCO and E 11 (4)HONiO pathways, respectively, due to inversion center located in both ring patterns.

a Chain and ring patterns of hydrogen bonds along (a) a axis and b b direction
Electronic spectra
The structural analysis of (1) shows a large angular distortion from the ideal octahedron. It was also confirmed with electronic spectra, measured in the solid state (diffuse reflectance) and water solution (absorbance).
Generally, hexacoordinate octahedral Ni(II) complexes show three broadbands in the NIR, visible and UV regions at 7000–13,000, 11,000–20,000, and 19,000–27,000 cm−1 assigned to the spin-allowed transitions: 3A2g → 3T2g (υ 1), 3A2g → 3T1g (F) (υ 2), and 3A2g → 3T1g (P) (υ 3), respectively [46]. These transitions are observed in the reflectance spectrum of (1) at 10,450 cm−1 (υ 1), 16,550 cm−1 (υ 2), and 25,850 cm−1 (υ 3). However, splitting of the highest energy d–d band (Fig. 5) indicates distortion of octahedral geometry. The digital filtration analysis of the spectrum revealed that the positions of the bands are typical for complexes of tetragonal (D4h) symmetry (Table 4; Fig. 6b). Therefore, further analysis of the solid-state spectrum was carried out assuming tetragonal geometry (vide infra).

Electronic spectra of [Ni(sar)2(H2O)2]: reflectance (solid line) and absorbance in water solution (dashed line) as received and upon digital filtration (inset)

a Correlation diagram (d8 configuration) for Oh and D4h symmetries and b the effect of filtration of the solid-state spectrum of [Ni(sar)2(H2O)2]
In contrast to the reflectance spectrum, band splitting was observed neither in the water solution spectrum nor in its filtered form (Fig. 5). The digital filtration revealed only bands of spin-forbidden transitions (3A2g → 1Eg = 13,400 cm−1, 3A2g → 1T2g = 21,400 cm−1). This indicates higher, i.e., pseudooctahedral, symmetry of (1) in solution [11].
On the basis of the band positions, crystal field and Racah B parameters for solid state and solution were calculated (Table 4). Obtained parameter values: Dq = 1016 cm−1 and B = 795 cm−1 of complex (1) in solution were close to the values found for pseudooctahedral Ni(II) complexes with [NiN2O4] chromophore [10, 11, 47, 48]. Crystal field Dq parameter in D4h symmetry has a slightly different meaning than that in Oh symmetry, because CF splitting in the former is described by three parameters: Dq, Ds, and Dt [49]. Thus, nephelauxetic parameter β, which means the ratio of the B value of complex to the B value of free ion (1041 cm−1 [46]), is the only one to be compared. A much higher value for the solid state (β = 0.94) was found than for solution (β = 0.76). This means that ionicity of M–L bond in solid state is significantly higher than in solution, which was observed earlier for other Ni(II) complexes [11].
On the basis of 10Dq (Δ) value obtained from solution spectra, sarcosine was located in the spectrochemical series of ligands for octahedral [NiL6] or pseudooctahedral [Ni(L–L)3] complexes. This series is constructed based on the crystal field parameter Δ for Oh symmetry and provides information about the strength of a ligand in the crystal field, i.e., its ability to split metal d levels in the electrostatic environment [46]. “Average environmental rule” [49] was applied in order to obtain the Δ value for hypothetical monoligand [Ni(sar)6]4− complex assuming exclusively monodentate N–Ni coordination. Δ was calculated from the equation:
Assuming Δ[Ni(H2O)6]2+ = 8500 cm−1 [46] and Δ[Ni(H2O)2L2] = 10,160 cm−1 (this work, Table 4), a hypothetical Δ[Ni(sar)6]4− was found to be 10,990 cm−1. This value locates sarcosine ligand in the spectrochemical series as follows: H2O (8500) < py (10,150) < NH3 (10,750) < 1-mhyd (10,750) < sar (10,990) < en (11,700) < bpy (12,650).
Analysis of known crystal structures of sarcosine metal complexes (see Sect. 3, Fig. 2) indicates that bidentate coordination [Ni(sar)3]− is more likely to occur. Moreover, sarcosine ligand in (1) is coordinated to Ni(II) ion with not only nitrogen atom (as it was assumed) but also with oxygen atom. Thus, the position of sarcosine in this row should be treated tentatively.
Geometry and charge distribution
The studied complex is an open-shell system, d8 electron configuration of Ni(II) cation with an pseudooctahedral environment of ligand donor atoms, which required the use of the unrestricted methods for the calculations of an electronic structure. The molecular parameters predicted for (1) are in good agreement with the X-ray diffraction results (Table 2). The calculated metal–ligand distances are equal to 2.002 (Ni1–O1), 2.224 Å (Ni1–O1 W), and 2.081 Å (Ni1–N1).
According to the NBO results, the electronic configuration of Ni is [core]4s(0.25)3d(8.32)4p(0.02): 18 core electrons, 8.59 valence electrons (on 4s and 3d atomic orbitals), and 0.02 Rydberg electrons, mainly on the 4p orbital. This gives the total number of 26.59 electrons, which is consistent with the calculated natural charge (+1.41) on the Ni atom in (1). The calculated natural charges on the atoms are collected in Table 5.
Vibrational spectra
The selected experimental and theoretical frequencies, IR intensities, and Raman intensities are shown in Table 6. All the experimental and calculated frequencies are listed in Table S1 of the Supporting Information. Figure 7 shows the experimental Raman and IR vibrational spectra of (1) in the range 4000–500 cm−1.

Experimental FT-IR and FT-Raman spectra in the range of 4000–500 cm−1
In the experimental Raman spectrum, the strong band is observed at 3247 cm−1. According to the PED calculation, this band is due to stretching vibration of NH group of sarcosine ligands. The subsequent Raman bands of strong intensities were assigned to the CH stretching of methyl and methylene groups of organic ligands. The characteristic band of symmetric CH stretching vibrations of N–CH3 group is observed at 2814 cm−1 in experimental Raman and IR spectra.
The characteristic band due to C=O stretching is observed at 1607 cm−1 in the experimental IR spectrum. This assignment is supported by the predicted large IR intensity of the mode 18.
According to the PED calculation, the strong band at 1397 cm−1 is associated with C–O stretching vibration. The separation of the bands due to CO stretching of carboxylic groups denoted as v as(COO−) and v s(COO−) over 200 cm−1 is consistent with monodentate manner of the coordination of sarcosine carboxylic group to the nickel ion.
The next strong IR band at 1320 cm−1 was assigned to the mode 33 with predominant contribution (66 %) from bending deformation of the CH2 groups.
The experimental Raman and IR spectra in the range of 500–100 cm−1 are presented in Fig. 8. As revealed by the PED calculation, the nickel–ligand vibrations are mixed with each other and also with different bending deformation of coordination rings of two sarcosine ligands. According to the calculated theoretical wave numbers, IR and Raman intensities, the bands due to vibrations with predominant contribution from v(Ni–O) and v(Ni–N) stretchings are observed at 445 cm−1 (IR) and 442 cm−1 (Raman).

Experimental FT-IR and FT-Raman spectra in the range of 500–100 cm−1
Summary and conclusions
Sarcosine complex [Ni(sar)2(H2O)2] was obtained by a novel route—as an unexpected solid product of the system: [Ni(II)–1-methylhydantoin]. Sarcosine was generated through basic hydrolysis of 1-methylhydantoin and was coordinated in situ to the metal ion. To the best of our knowledge, this is the first example of the sarcosine complex synthesis, which starts from a substrate other than sarcosine itself. Moreover, the crystals of the complex are of better quality than those obtained with the traditional method. Although the compound was known earlier, the hydrogen bond analysis and spectroscopic (IR, Raman, and NIR–UV–Vis) studies combined with DFT calculations presented here allowed for its better characterization. Single crystal X-ray diffraction revealed that adjacent molecules are bound by H-bonds. It leads to the formation of a 2D network, created by two chain patterns along the main crystallographic directions a and b. Solid-state reflectance electronic spectrum strengthened by digital filtration proved that geometry of the nickel(II) surrounding can be described as tetragonal. However, in water solution, the complex exhibits higher, pseudooctahedral symmetry. Crystal field and Racah B parameters were calculated for both tetragonal and pseudooctahedral geometries. Comparison of nephelauxetic parameters calculated for (1) in solid state and solution shows that M–L bonds have a more ionic nature in the solid state than in the water solution. By application of the “average environment rule,” sarcosine was tentatively ranked in the spectrochemical series of ligands. The obtained 10Dq value of 10,990 cm−1 located sarcosine between ammonia and 1-methylhydantoin. It means that this amino acid has moderately strong splitting ability.
The PED analysis revealed that carboxylic groups of sarcosine ligands show a monodentate mode of coordination to the nickel atom. The presence of the Ni1–N1 and Ni1–O1 bands in the vibrational spectra of the studied complex is consistent with the presence of the chelating rings formed during coordination of nickel by sarcosine ligands.
References
Mudd SH, Ebert MH, Scriver CR (1980) Metabolism 29:707
Katz E, Goss WA (1959) Biochem J Nov 73:458
Lloyd AW, Baker JA, Smith G, Olliff CJ, Rutt KJ (1992) J Pharm Pharmacol 44:507
Strzelecki D, Szyburska J, Rabe-Jabłońska J (2014) Neuropsych Dis Treat 10:263
Ashida T, Bando S, Kakudo M (1972) Acta Cryst B28:1560
Krishnakumar RV, Natarajan S (1995) Cryst Res Technol 30:825
Krishnakumar RV, Subha Nadhini M, Natarajan S (2001) Acta Cryst 57:192
Gawryszewska PP, Jerzykiewicz L, Sobota P, Legendziewicz J (2000) J Alloys Compd 300:275
Sabo TJ, Dinović VM, Kaluderović GN, Stanojković TP, Bogdanović GA, Juranić ZD (2005) Inorg Chim Acta 358:2239
Puszyńska-Tuszkanow M, Daszkiewicz M, Maciejewska G, Adach A, Cieślak-Golonka M (2010) Struct Chem 21:315
Puszyńska-Tuszkanow M, Daszkiewicz M, Maciejewska G, Staszak Z, Wietrzyk J, Filip B, Cieślak-Golonka M (2011) Polyhedron 30:2016
Puszyńska-Tuszkanow M, Grabowski T, Daszkiewicz M, Wietrzyk J, Filip B, Maciejewska G, Cieślak-Golonka M (2011) J Inorg Biochem 105:17
Puszyńska-Tuszkanow M, Staszak Z, Misiaszek T, Klepka MT, Wolska A, Drzewiecka-Antonik A, Fałtynowicz H, Cieślak-Golonka M (2014) Chem Phys Lett 597:94
Ware E (1950) Chem Rev 46:403
Puszyńska-Tuszkanow M, Daszkiewicz M, Maciejewska G, Cieślak-Golonka M (2009) Inorg Chem Commun 12:484
Guha S (1973) Acta Cryst B29:2167
Sheldrick GM (2008) Acta Cryst A64:112
Hübschle CB, Sheldrick GM, Dittrich B (2011) J Appl Cryst 44:1281
Biermann G, Ziegler H (1986) Anal Chem 58:536
Myrczek J (1990) Spectr Lett 23:1027
Underhill AE, Billing DE (1966) Nature 210:834
Bartecki A, Kurzak K (1981) Bull de L’Academie Polonaise des Sci 29:299
Perdew JP, Burke K, Ernzerhof M (1996) Phys Rev Lett 77:3865
Ernzerhof M, Scuseria GE (1999) J Chem Phys 110:5029
Adamo C, Barone V (1999) J Chem Phys 110:6158
Hay PJ, Wadt WR (1985) J Chem Phys 82:270
Menon AS, Radom L (2008) J Phys Chem A 112:13225
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian Inc., Wallingford CT
Reed AE, Curtiss LA, Reinhold F (1988) Chem Rev 88:899
Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Weinhold F (2001) NBO 5.0 software. Theoretical Chemistry Institute, University of Wisconsin, Madison
Nowak MJ, Lapinski L, Bieńko DC, Michalska D (1997) Spectrochim Acta A 53:855
Rostkowska H, Lapinski L, Nowak MJ (2009) Vib Spectrosc 49:43
Fogarasi G, Zhou X, Taylor PW, Pulay P (1992) J Am Chem Soc 114:8191
Lapinski L., Nowak M.J., unpublished computer program BALGA for PED calculations
Polavarapu PL (1990) J Phys Chem 94:8106
Keresztury G, Holly S, Besenyei G, Varga J, Aiying Wang, Durig JR (1993) Spectrochim Acta A 49:2007
Michalska D, Wysokiński R (2005) Chem Phys Lett 403:211
Krishnakumar RV, Natarajan S, Bahadur SA, Cameron TS (1994) Z Kristallogr 209:443
Inomata Y, Shibata A, Yukawa Y, Takeuchi T, Moriwaki T (1988) Spectrochim Acta A 44:97
Larsen S, Watson KJ, Sargeson KM, Turnbull KR (1968) Chem Commun 15:847
Trzebiatowska-Gusowska M, Gągor A, Baran J, Drozd M (2009) J Raman Spectrosc 40:315
Silva MR, Beja AM, Paixao JA, de Veiga LA (2001) Z Kristallogr New Cryst Struct 216:419
Fleck M, Ghazaryan VV, Petrosyan AM (2013) Acta Cryst C69:11
Etter MC, MacDonald JC, Bernstein J (1990) Acta Cryst B46:256
Daszkiewicz M (2012) Struct Chem 23:307
Lever ABP (1984) Inorganic Electronic Spectroscopy. Elsevier, New York
König E (1971) Struct Bond 9:175
Małecki JG, Mroziński J, Michalik K (2011) Polyhedron 30:1806
Lever ABP, Nelson SM, Shepherd TM (1965) Inorg Chem 4:810
Acknowledgments
The authors are grateful to Dr. Mariola Puszyńska-Tuszkanow for her helpful advice during the practical part of this work. Mrs. Elżbieta Mróź and M.Sc. Magdalena Malik are acknowledged for their measurements of the IR and Raman spectra, respectively. This work was financed in part by a statutory activity subsidy from the Polish Ministry of Science and Higher Education for the Faculty of Chemistry of Wroclaw University of Technology. The generous computer time from the Wroclaw Supercomputer and Networking Center and the Poznan Supercomputer and Networking Center is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor Magdolna Hargittai on the occasion of her 70th birthday.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fałtynowicz, H., Daszkiewicz, M., Wysokiński, R. et al. Ni(II) complex with sarcosine derived from in situ generated ligand: structural, spectroscopic, and DFT studies. Struct Chem 26, 1555–1563 (2015). https://doi.org/10.1007/s11224-015-0631-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-015-0631-7