
Abstract
DFT calculation results shed a new light on the mechanism of cycloaddition reaction between tetrafluoroethene and cyclopentadiene. The unique influence of fluorine atoms on the ethylene derivative molecule causes the [2 + 2] cycloaddition process to take place according to a stepwise, biradical mechanism. At the same time, the competitive and independent path leads to a one-step (and not a two-step, as was once thought) cycloaddition reaction leading to a [2 + 4] cycloadduct.
Similar content being viewed by others
Introduction
The Diels–Alder (DA) reaction is the most universal and flexible method of synthesis of six-membered carbo- and heterocyclic compounds [1–4]. It used to be assumed that these reactions—regardless of adduct structure—take place according to a one-step, “pericyclic” mechanism. However, the most recent studies undermine this view. In some cases, it was proven that DA reactions may actually take place according to a two-step mechanism [5–10]. Such a two-step mechanism with biradical intermediate takes place e.g. in the cycloaddition reaction of 1,2-dichloroethene with hexachlorocyclopentadiene [5], and 2-chlorobuta-1,3-diene dimerization reaction [6]. On the other hand, reactions e.g. of 3-nitropyridine with 1-methoxy-3-trimethoxysillylbuta-1,3-diene [7], 4,6-dinitrobenzofuroxan with 1-trimethylsilyloxybuta-1,3-diene [8], aryl-substituted 1,2,4-triazines with 2-cyclopropylidene-1,3-dimethylimidazolidine [9] and 1,1,1-trifluor-3-nitroprop-2-ene derivatives with enamines [10] take place according to a two-step, zwitterionic mechanism.
It was determined empirically [11, 12] some time ago that the cycloaddition reaction between tetrafluoroethene (1) and cyclopentadiene (2) result in a [2 + 2] adduct (4) in addition to the expected tetrafluoronorbornene (3) (Scheme 1). The presence of the former compound in the final reaction mass inclined the authors of the work [11] to propose that the analyzed reaction takes place according to a two-step, biradical mechanism. This is possible, because the [2 + 2] type cycloaddition reactions do not take place as one-step processes. It was also known [13], that radical centers of intermediates may be stabilized in the direct vicinity of fluorine atoms.

Hypothetical biradical mechanism for cycloaddition reaction between tetrafluoroethene 1 and cyclopentadiene 2
According to the authors of work [11], a biradical intermediate IN is formed in the first step of the reaction, which may later undergo cyclisation to a [2 + 4] or [2 + 2] cycloadduct along competitive paths. Unfortunately, the two-step mechanism of the analyzed cycloaddition reaction could not be confirmed. Also, the final nature of the intermediate has not been decisively proven. With the aforementioned issues in mind, this work was designed in order to perform quantum-chemical studies of reaction between tetrafluoroethene and cyclopentadiene. The following studies were performed, in particular: (i) an analysis regarding the nature of adduct interactions in the elementary reaction act based on the recently developed [14–19] theory of reactivity indices, and (ii) detailed simulations of theoretically possible conversion paths of substrates into products were performed.
Calculation methods
All calculations reported in this paper were performed on “Zeus” supercomputer in the “Cyfronet” computational centre in Cracow. Hybrid functional B3LYP with the 6-311G(d), basis set included in the GAUSSIAN 09 package [20] was used. Analogical functional has been used for analysis of mechanistic problems of several processes involving fluoroorganic compounds—cycloadditions [21–23] as well as other reactions [24, 25].
Global electronic properties of reactants were estimated according to the equations defined on the basis of conceptual density functional theory [16, 26]. According to Domingo suggestions [27] for these calculations, B3LYP/6-31G(d) theory level was used. In particular, the electronic chemical potentials (μ) and chemical hardness (η) were evaluated in terms of one-electron energies of FMO (EHOMO and ELUMO) using the following equations:
Next, the values of μ and η were then used for the calculation of global electrophilicity (ω) according to the formula:
Subsequently, global nucleophilicities (N) [28] of 1 and 2 can be expressed in terms of equation:
The results are collected in Table 1.
The stationary points on reaction paths were localized in an analogous manner as in the case of the previously analyzed DA reaction of conjugated nitroalkenes with alkyl-vinyl ethers [29, 30]. In particular, for the structure optimization of the reactants and the reaction products, the Berny algorithm [31] was applied. First order saddle points were localized using the QST2 procedure. Stationary points were characterized by frequency calculations. All reactants, products, and local minima had positive Hessian matrices. All transition states showed only one negative eigenvalue in their diagonalized Hessian matrices, and their associated eigenvectors were confirmed to correspond to the motion along the reaction coordinate under consideration. IRC calculations were performed to connect previously computed transition structures with suitable minima. For optimized structures, the thermochemical data for the temperature T = 748 K and pressure p = 1 atm were computed using vibrational analysis data. Carbon kinetic isotope effects on reaction centers were calculated on the basis of values of rate constants calculated for reactions involving “non-marked” (kC12) and “13C-marked” (kC13) compounds, according to the formula:
For this purpose, rate constants which were computed using classical Eyring equation was applied.
The kinetic parameters as well as essential properties of critical structures are displayed in Tables 2, 3 and 4.
Results and discussion
Nature of interaction between addents
First and foremost, it was decided to diagnose the nature of interactions between reacting molecules in the elementary reaction act. The theory of global electrophilicity and nucleophilicity indices [26, 27] was employed in this task. This approach was recently and successfully used in interpretation of the course of various [2 + 2] and [2 + 4] cycloaddition reactions [7, 29, 30, 32–34].
As can be concluded from data provided in Table 1, both reaction components are characterized by low global electrophilicity ω, which does not exceed 1 eV. In particular, electrophilicity of 1 is equal to only 0.6 eV, thus the process should be considered to be very poor polar. In addition, the very low value of Δμ is in agreement with the low polar character of the reaction. In consequence, this process, similarly as DA reaction between cyclopentadiene and ethylene, according to Domingo classification [35] may be considered as non-polar Diels–Alder reaction (N-DA). The small Δω value indicates that the “driving force” of the cycloaddition process is small, which explains why the reaction takes place under relatively difficult conditions in practice. The maximum charge which the ethylene derivative may accept in the transition state (ΔNmax) is almost 0.4e.
Kinetic aspects
RB3LYP/6-311G(d) calculations indicate that the conversion of reagents into the [2 + 4] cycloadduct (3) takes place via a single activation barrier, which requires significant Gibbs free energy expense to be overcome (about 230 kJ/mol) (Table 2; Fig. 1). This indicates a one-step reaction mechanism. An alternative, kinetically allowed path of reagent conversion ultimately leads to a [2 + 2] adduct 4. In this case, two transition states have been located between the substrate and the product minimums, separated by an intermediate well (Fig. 1). In the first step, the reacting system reaches a maximum related to reaching the TS-B transition state, which requires a Gibbs free energy expense about 240 kJ/mol.

Energy profiles for cycloaddition between tetrafluoroethene 1 and cyclopentadiene 2 in gas phase and solution according to B3LYP/6-311G(d) calculations
Further shifting of the reacting system along the reaction coordinate leads to a thermodynamically unstable intermediate IN, which undergoes conversion via a relatively small activation barrier (ΔG = 48.7 kJ/mol) to the final product 4. All attempts aimed at finding an alternative path leading to the adduct 5 in a single step failed. No path leading directly from intermediate INto the [2 + 4] cycloadduct 3 exists, either. The INto 3 conversion may only proceed via a dissociation stage into individual reagents, followed by a single-step 1 + 2 → 3 cycloaddition.
Thus, B3LYP/6-311G(d) calculations clearly indicated that contrary to earlier suggestions [11, 12] (Scheme 1), the acyclic adduct 3 is not a common intermediate for [2 + 2] and [2 + 4] cycloaddition reactions.
Key structures
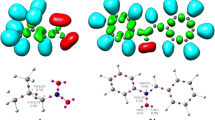
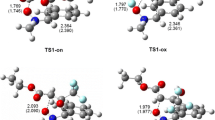
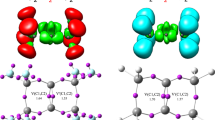
First, an analysis of key properties of critical structures obtained using the B3LYP/6-311G(d) calculations was performed. It turned out that the TS-A transition state is a two-plane structure (Fig. 2), typical for one-step DA reactions between cyclopentadiene and ethylene derivatives [36–39]. Both new σ-bonds are formed simultaneously within this structure. However, these bonds are not significantly advanced (r > 2.25 Å) (Table 3). The formation of new bonds is accompanied by sp2 → sp3 rehybridization at the reaction centers (Table 4). In the case of C1 and C4 cyclopentadiene centers, however, the sp2 → sp3 rehybridization is very poor. This fact is indicated by the values of carbon kinetic isotope effects (KIE), which do not reach 1.010. Slightly more profound isotopic effects indicate a slightly more significant sp2 → sp3 rehybridization of reaction centers of the ethylene derivative. In general, however, TS-A should be classified as a so-called early transition state, in which reaction center hybridization resembles the initial sp2 hybridization, observed in substrates, much more closely than sp3 hybridization observed in products. The value of the GEDT [15] index (0.03e) also indicate that the analyzed transition state is weakly polar.

Views of key structures for cycloaddition between tetrafluoroethene 1 and cyclopentadiene 2 in gas phase
The first critical structure present along the path of reagent conversion into the cyclobutane derivative 4 is provided by the TS-B transition state. Only one new σ-bond is formed in this state (Fig. 2). This is the bond between the C1 atom of cyclopentadiene 2 and the C5 carbon atom of tetrafluoroethene 1. It reaches the length of 1.850Å within TS-B. Analysis of carbon kinetic isotope effects at reaction centers of the transition state (Table 4) leads to interesting conclusions. This suggests that sp2 → sp3 rehybridization within TS-B takes place not only at the C1 and C5 centers, but also at the C6 atom (which does not participate in formation of any new bonds at this reaction stage). On the other hand and according to the expectation, isotope effects at C2 and C4 centers (introduced by cyclopentadiene) are practically 1, which means that no practically significant rehybridization takes place at these atoms within TS-B. It should be noted, that the value of the GEDT [15] index (0.09e) indicate moderately polar nature of this transition state.
Another critical structure along the path of reagent conversion into compound 4 includes the acyclic intermediate IN. The C1-C5 bond is already formed in this intermediate. Reoptimization of IN at UB3LYP/6-311G(d) theory level yields a structure which is more stable than the close-shell configuration. This observation confirmed the biradical character of IN.
The third critical structure along the path of adduct conversion into cycloadduct 4 includes the TS-C transition state. The second σ bond required in order to form a cyclobutane ring forms in this transition state. This is the C2-C6 bond, which reaches 3.106 Å at this stage. Thus, this bond is very weakly formed. This is confirmed by the values of isotope effects at C2 and C6 atoms, which only slightly exceed 1. Similarly to the case of IN, UB3LYP/6-311G(d) calculations suggest biradicaloidal nature of TS-C.
Further shift of the reacting system from the TS-C state along the reaction coordinate leads to the formed product 4. In a summary, the mechanism of cycloaddition reaction of tetrafluoroethene 1 with cyclopentadiene 2 should be interpreted as shown in Scheme 2.

Mechanism of cycloaddition reaction between tetrafluoroethene 1 and cyclopentadiene 2 according to B3LYP/6-311G(d) calculations
Conclusion
DFT calculation data shed a new light on the mechanism of cycloaddition reaction between tetrafluoroethene and cyclopentadiene. Contrary to the previous views, the acyclic intermediate formed along the path of reagent conversion is not a common transition product for paths of [2 + 4] and [2 + 2] cycloaddition. The only possible path of its cyclisation includes the path leading to a cyclobutane derivative. The Diels–Alder adduct is formed in a competitive, one-step reaction.
References
Funel J-A, Abele S (2013) Angew Chem Int Ed 52:3822–3863
Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G (2002) Angew Chem Int Ed 41:1668–1698
Dresler E, Jasińska E, Łapczuk-Krygier A, Nowakowska-Bogdan E, Jasiński R (2015) Chemik 69:288–296
Juhl M, Tanner D (2009) Chem Soc Rev 38:2983–2992
Bolduc PR, Goe GL (1974) J Org Chem 39:3179–3181
Firestone RA (1996) Tetrahedron 52:14459–14468
Ormachea CM, Mancini PME, Kneeteman MN, Domingo LR (2015) Comp Theor Chem 1072:37–42
Lakhdar S, Terrier F, Vichard D, Berionni G, El-Guesmi N, Goumont R, Boubaker T (2010) Chem Eur J 16:5681–5690
Ernd M, Heuschmann M, Zipse H (2005) Helv Chim Acta 88:1491–1518
Korotayev VYU, Barkov AYU, Slepukhin PA, Kodess MI, Sosnovskikh VYA (2011) Mendeleev Commun 21:112–114
Drysdale J, Gilbert WW, Sinclair HK, Sharkey WH (1958) J Am Chem Soc 80:3672–3675
Christova NB, Pavlova SD, Kostov GK (1993) React Kinet Catal Lett 49:393–402
Huisgen R, Giera H, Polborn K (2005) Tetrahedron 61:6143–6153
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Tetrahedron 58:4417–4423
Domingo LR (2014) RSC Adv 4:32415–32428
Geerlings P, De Proft F, Langenaeker W (2003) Chem Rev 103:1793–1873
Kapłon K, Demchuk OM, Wieczorek M, Pietrusiewicz KM (2014) Current Chem Lett 3:23–36
Jasiński R, Ziółkowska M, Demchuk OM, Maziarka A (2014) Central Eur J Chem 12:586–593
Jasiński R (2015) Monatsh Chem 146:591–599
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09. Gaussian Inc, Wallingford
Jasiński R (2015) J Fluor Chem 176:35–39
Rahimizadeh M, Eshghi H, Khojastehnezhad A, Moeinpour F, Bakavoli M, Tajabadi J (2014) J Fluor Chem 162:60–65
Jasiński R, Socha J, Barański A (2013) Chem Heterocyclic Compd 49:1055–1060
Łapczuk-Krygier A, Korotaev VYU, Barkov AYU, Sosnovskikh VYU, Jasińska E, asiński R (2014) J Fluor Chem 168:236–239
Jasiński R (2014) J Fluor Chem 160:29–33
Parr RG, Yang W (1989) Density functional theory of atoms and molecules. Oxford University, New York
Perez P, Domingo LR, Aurell MJ, Contreras R (2003) Tetrahedron 59:3117–3125
Domingo LR, Chamorro E, Perez P (2008) J Org Chem 73:4615–4624
Jasiński R (2014) Comp Theor Chem 1046:93–98
Jasiński R, Kubik M, Łapczuk-Krygier A, Kącka A, Dresler E, Boguszewska-Czubara A (2014) Reac Kinet Mech Cat 113:333–345
Hratchian HP, Schlegel B (2005) In: Dykstra C (ed) Theory and applications of computational chemistry. Elsevier, Amsterdam
Domingo LR, Ríos-Gutiérrez M, Pérez P (2015) Tetrahedron 71:2421–2427
Emamian S, Hosseini SJ, Ravani K-S (2015) RSC Adv 5:98538–98548
Jasiński R (2016) Monatsh Chem. doi:10.1007/s00706-016-1735-5 in Press
Domingo LR, Chamorro E, Sáez JA (2010) Org Biomol Chem 8:5495–5504
Jasiński R, Kwiatkowska M, Sharnin V, Barański A (2013) Monatsh Chem 144:327–335
Jasiński R, Kwiatkowska M, Barański A (2009) J Mol Struct (Theochem) 910:80–87
Jasiński R, Kwiatkowska M, Barański A (2011) J Phys Org Chem 24:843–853
Kwiatkowska M, Jasiński R, Mikulska M, Barański A (2010) Monatsh Chem 141:545–548
Acknowledgments
The regional computer center “Cyfronet” in Cracow (Grant No. MNiSW/Zeus_lokalnie/PK/009/2013) is thanked for the allocation of computing time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jasiński, R. A reexamination of the molecular mechanism of the Diels–Alder reaction between tetrafluoroethene and cyclopentadiene. Reac Kinet Mech Cat 119, 49–57 (2016). https://doi.org/10.1007/s11144-016-1038-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-016-1038-1