
Abstract
Kinetic measurements show that despite their polar nature, cycloaddition reactions of (E)-3,3,3-trichloro-1-nitroprop-1-ene with ketonitrones take place according to a mechanism that proceeds without intervention of zwitterionic intermediate. This is indicated by the values of activation parameters (activation enthalpy, activation entropy) and the nature of the kinetic solvent effect.
Similar content being viewed by others

Introduction
The mechanistic aspects of [3+2] cycloaddition reactions have recently drawn ever greater attention from organic chemists [1–10]. It turned out that in the case of reactions including strongly electrophilic dipolarophiles, a stepwise, zwitterionic mechanism may compete with a classical one-step mechanism. In recent years, it has been proven that such a mechanism takes place e.g. in the case of [3+2] cycloadditions between N-methylnitrone and fluorinated alkenes [6] as well as between azomethine ylides and dialkyl-2,3-dicyanobut-2-enedioates [10]. The stepwise, zwitterionic mechanism of [3+2] cycloaddition may also be enforced by the presence of ionic liquids in the reaction medium [3].
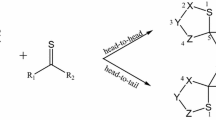
This work is a continuation of our complex studies [3, 5, 11–14] on the stereochemistry and mechanistic aspects of [3+2] cycloaddition reactions. We have previously [11] determined that reactions of (E)-3,3,3,-trichloro-1-nitroprop-1-ene (1) with ketonitrones take place under relatively mild conditions and lead to adducts with a nitro group at position C4 of the isoxazolidine ring (Scheme 1).

Reaction course of the 1,3-dipolar cycloaddition between (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and ketonitrones 2a,b
However, the matter of the mechanism of these reactions remains to be solved. On the one hand, we have shown that these reactions take place with retention of the original substituents configuration in the dipolarophile molecule. This is rather characteristic of one-step cycloaddition reactions [15, 16]. On the other hand, it is probable that this process takes place according to a stepwise mechanism, but products with 4,5-cis configuration are not created because the rotation barrier within the zwitterionic intermediate (path B) is significantly higher than the energy required to close the ring with retention of the original substituents configuration (path A) (Scheme 2).

Hypothetical stepwise mechanism for [3+2] cycloaddition between (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and ketonitrones 2a,b
Of importance is also the fact that because of the large steric crowding of substituents at the C4 and C5 atoms, steric factors do not favor the thermodynamic stability of compounds I 2 and 4. On the other hand, an analysis of electrophile–nucleophile interactions performed on the basis of the analysis of the conceptual DFT indices (extensively developed in recent years [17–22]) showed [11] that the studied processes are of a clearly polar nature and electronic factors favor fast bond formation between the nitrone oxygen atom and the β-carbon atom of the dipolarophile. In addition, DFT simulations [11] of reaction paths indicate an extremely asynchronous nature of transition states (TSs). Thus, the determination of the mechanism of the analyzed reactions as well as diagnosing factors stimulating their course requires the performance of further, more thorough experimental studies. Taking the above into account, we have undertaken kinetic studies of model reactions using (E)-3,3,3-trichloro-1-nitroprop-1-ene (1), C,C,N-triphenylnitrone (2a) and C-fluorene-N-phenylnitrone (2b) as a part of this work In particular, we have decided to: (i) perform activation parameters measurements, and (ii) study kinetic solvent effects.
Experimental procedure
The rates of cycloaddition were followed by measuring the area of HPLC peak of the nitrone. The reactions were initiated by introducing a weighed quantity of nitrone to the stirred and thermostated solution (50 mL) of the nitroalkene in a suitable solvent. The initial concentration of the former was 0.007 mol L−1, whereas the latter was always in 12-fold molar excess. The kinetic runs were carried out at temperatures as indicated in Table 1 up to 80 % completion. In this way, pseudo-first order rate constants were measured according to the following equation:
In this equation, [N] is the nitrone concentration, k* is the pseudo-first order rate constant.
The second order rate constants (k) were obtained from the quotient of k* and initial concentration of nitroalkene ([A]):
For reaction testing, a Knauer apparatus equipped with UV–VIS detector, and Lichrospher 100-10 RP18 column (4 × 250) was applied. Methanol–water mixtures (70:30 v/v) were used as eluent at the flow rate 1.2 mL min−1. The results of kinetic measurements are collected in Table 1. On the basis of the rate constants measured at different temperatures, the activation enthalpy and activation entropy were calculated, using the Eyring equation [16]:
Standard errors has been calculated according to approach proposed by Lente et al. [23, 24].
Nitroalkene and nitrones were prepared according to the methods described in the literature: 1—[25, 26], 2a,b—[27].
Results and discussion
As can be concluded from the measurements, the reaction of E-3,3,3-trichloro-1-nitroprop-1-ene 1 with triphenylnitrone 2a in a toluene solution takes place relatively fast, already at room temperature (k = 73.74 L mol−1 s−1). For comparison, we have performed a test of a similar reaction using nitroethene (5), under identical conditions. It turned out, surprisingly, that this process takes place almost three times more slowly. It can be concluded from the above that not steric, but electronic factors have the decisive influence on the course of these reactions. Nitroethene has a significantly less strongly shielded C=C bond, but at the same time it has clearly lower global electrophilicity (ω = 2.61 eV [17], in comparison to 3.43 eV in case of E-3,3,3-trichloro-1-nitroprop-1-ene [14] ).
However, detailed tests of the reaction using nitrone 2b have shown that in order to achieve a similar order of magnitude of the reaction constant, it is necessary to increase the temperature up to 70 °C. The phenomenon of relatively lower reactivity of fluorenonitrone 2b in comparison with triphenylnitrone 2a, however, may be explained. The 2b molecule shows a tendency to retain the sp2 configuration of the carbon atom of the >C=N(O)– group caused by the conjugation of the pz orbital of this atom with pz orbitals of the fluorene system. They lie in the same plane and such interactions are fully possible. In the case of triphenylnitrone, there are no similar, stabilizing interactions, thus the sp2–sp3 rehybridization may progress much faster in the course of the reaction.
Next, we have performed a series of measurements for a 1+2a and 1+2b reactions at various temperatures. On the basis of these measurements, we have determined activation parameters, using the Eyring equation.
It turned out that activation enthalpies of the analysed reactions do not exceed tens kJ mol−1 (respectively, 42.26 ± 1.05 and 54.15 ± 1.62 kJ mol−1 for reactions 1 + 2a and 1 + 2b). For comparison, in the case of the one-step cycloaddition of (Z)-C,N-diphenylnitrone with dimethyl fumarate, the activation enthalpy is 54.4 kJ mol−1 [28].
In turn, activation entropies have large, negative values (−144.11 ± 3.6 and −147.11 ± 4.4 J mol−1 K−1 for reactions 1 + 2a and 1 + 2b). This is, in turn, characteristic of reactions taking place via strongly stiffened transition states. Thus, e.g., in the case of the evidently one-step cycloaddition of 1-aza-1-cyclooctene-1-oxide with methyl acrylate, the activation entropy is −131 J mol−1 K−1, whilst in the case of a similar reaction with methyl methacrylate, it is −148 J mol−1 K−1 [29].
This means that measurements of activation parameters indicate a rather strongly ordered nature of the transition states, thus also rather a one-step mechanism of the studied cycloaddition reactions. In order to clearly determine whether zwitterionic intermediates will appear in the reaction medium, we have performed studies on solvent influence on reaction kinetics.
Thus, it turned out that the reaction of E-3,3,3-trichloro-1-nitroprop-1-ene 1 with triphenylnitrone 2a in weakly polar solvents takes place faster than in strongly polar solvents (Table 1). In particular, this reaction constant in CCl4 is about 50 % higher than in nitromethane. A correlation analysis showed the existence of a linear relationship between log k values and values of Dimroth–Reinchardt constants [30] (Fig. 1). This relationship is quantitatively described using the following correlation equation:

Plot of log k vs Dimroth ET(30) constants for [3+2] cycloaddition between (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and C,C,N-triphenylnitrone 2a (25 °C)
The minus sign in the equation means that the transition state of the reaction is less polar than the substrates (the mass of which is dominated by the polar nitroalkene). The relatively low absolute value of the reaction constant means that the sensitivity of the process to medium polarity is rather weak and characteristic of processes that proceed without intervention of zwitterionic intermediate.
We have also performed similar studies for cycloaddition reaction of E-3,3,3-trichloro-1-nitroprop-1-ene 1 with nitrone 2b. It turned out that in this case, the value of the reaction constant increases as polarity of reaction medium increases. It is quantitatively described by the correlation equation:
The small absolute value of reaction constant also indicates a mechanism that proceeds without the intervention of a zwitterionic intermediate. However, the positive sign of this constant proves that the transitions state of this reaction has a much stronger polar nature than the substrates (Fig. 2).

Plot of log k versus Dimroth ET(30) constants for [3+2] cycloaddition between (E)-3,3,3-trichloro-1-nitroprop-1-ene (1) and C-fluorene-N-phenylnitrone 2b (70 °C)
Conclusions
Despite the fact that the nature of interaction between E-3,3,3-trichloro-1-nitroprop-1-ene with ketonitrones does not raise doubts, kinetic studies indicate that the aforementioned cycloadditions do take place according to mechanism. The small absolute value of the rate constant indicates a mechanism that proceeds without the intervention of a zwitterionic intermediate. This is confirmed by the values of activation parameters and the influence of solvent polarity on process kinetics. These studies also indicate that the more asynchronous and clearly more polar nature will be found in cycloaddition using C-fluorenonitrone, in the molecules of which electronic factors slow the rehybridization process on the carbon atom of the >C=N(O)– group.
References
Mlostoń G, Urbaniak K, Linden A, Heimgartner H (2015) Helv Chim Acta 98:453–461
Mlostoń G, Pipiak P, Linden A, Heimgartner H (2015) Helv Chim Acta 98:462–473
Jasiński R (2015) Tetrahedron Lett 56:532–535
Khlebnikov AF, Koneva AS, Virtseva AA, Yufit DS, Mlostoń G, Heimgartner H (2014) Helv Chim Acta 97:453–470
Jasiński R (2013) Tetrahedron 69:927–932
Wójtowicz-Rajchel H, Koroniak H (2012) J Fluor Chem 135:225–230
Krompiec S, Bujak P, Malarz J, Krompiec M, Skórka Ł, Pluta T, Danikiewicz W, Kania M, Kusz J (2012) Tetrahedron 68:6018–6031
Lan Yu (2010) Houk KN. J Am Chem Soc 132:17921–17927
Ess DH, Houk KN (2008) J Am Chem Soc 130:10187–10198
Huisgen R, Pöchlauer P, Młostoń G, Polsborn K (2007) Helv Chim Acta 90:983–998
Jasiński R, Mróz K, Kącka A (2015) J Heterocycl Chem 21:1–7
Jasiński R (2015) Monatsh Chem 146:591–599
Jasiński R (2015) J Heterocycl Chem 52:185–192
Jasiński R, Ziółkowska M, Demchuk OM, Maziarka A (2014) Cent Eur J Chem 12:586–593
Huisgen R (1984) In: Padwa A (ed) 1,3-Dipolar cycloaddition chemistry. Wiley Interscience, New York
Schwetlick K (1971) Kinetische Metoden zur Untersuchung von Reaktionsmechanismen. VEB, Berlin
Domingo LR, Aurell MJ, Perez P, Contreras R (2002) Tetrahedron 58:4417–4423
Perez P, Domingo LR, Aizman A, Contreras R (2007) In: Toro-Labbe´ A (ed) Theoretical aspects of chemical reactivity, vol 19. Elsevier, Amsterdam
Chattaraj PK, Giri G, Duley S (2011) Chem Rev 111:43–75
Domingo LR, Perez P, Saez JA (2013) RSC Adv 3:1486–1494
Kapłon K, Demchuk OM, Wieczorek M, Pietrusiewicz KM (2014) Current Chem Lett 3:23–36
Benchouk W, Mekelleche SM (2015) RSC Adv 5:22126–22134
Lente G, Fábián I, Poe AJ (2005) New J Chem 29:759–760
Lente G (2015) Deterministic kinetics in chemistry and systems biology the dynamics of complex reaction networks. Springer, New York
Compton M, Higgins H, MacBeth L, Osborn J, Burkett H (1949) J Am Chem Soc 71:3229–3231
Jasiński R (2013) Preparatyka alifatycznych nitrozwiązków. RTN, Radom
Staudinger H, Mischer K (1919) Helv Chim Acta 2:554–582
Gholami MR, Yangjeh AH (2000) Int J Chem Kinet 32:431–434
Al-Jaroudi SS, Perzanowski HP, Wazeer MIM, Ali SA (1997) Tetrahedron 53:5581–5592
Reichardt Ch, Welton T (2011) Solvents and solvent effects in organic chemistry. Wiley-VCH, Weinheim
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jasiński, R., Mróz, K. Kinetic aspects of [3+2] cycloaddition reactions between (E)-3,3,3-trichloro-1-nitroprop-1-ene and ketonitrones. Reac Kinet Mech Cat 116, 35–41 (2015). https://doi.org/10.1007/s11144-015-0882-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-015-0882-8