
Abstract
A series of CuO/CZ catalysts with different amounts of copper oxide were synthesized from copper nitrates by incipient wetness impregnation of the ceria–zirconia support. The structural, redox, acidic and adsorption properties were investigated using X-ray diffraction (XRD), BET, H2 temperature programmed reduction (H2-TPR), temperature programmed desorption of NO in the presence of oxygen (NO-TPD) and in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS). The results have shown good catalytic activity for CuO/Ce0,62Zr0,38O2 in NO oxidation to NO2. Species such as nitrites and nitrates were formed on the surface. The transformation of nitrite compounds into nitrate was observed above 200 °C. From 300 up to 400 °C, decomposition of the surface nitrate to nitrite was discerned. Ammonia was adsorbed on the Lewis and Brønsted acid sites on Cu/CZ catalyst surface during temperature programmed reactions. The evolution of adsorbed NH3 species to NH2 surface group was confirmed by formation specific band of absorption at 1528 cm−1.
Similar content being viewed by others


Introduction
The exhaust gases from stationary and mobile combustion sources containing, among others, nitrogen oxides, i.e. NO and NO2, are a major source of air pollution. These nitrogen oxides lead to photochemical smog, acid rain and ozone depletion [1, 2]. In order to decrease the NOx emissions according to the environmental emission standards, a various catalytic methods have been developed, e.g. the three-way catalysts for gasoline fuelled vehicles, NOx storage-reduction systems for lean burn engines or the selective catalytic reduction (SCR) processes for the large-scale combustion facilities [3–5]. The selective catalytic reduction of NOx has been widely studied. The key roles in this process are played by both the catalyst and the reducing agent, which can be ammonia, urea or hydrocarbons. Ammonia is considered to be the most active in the selective catalytic reduction of NOx (SCR-NH3) [10].
Various catalysts such as zeolites, metal oxides and noble metal catalysts have been widely investigated in the deNOx process [1, 4, 5]. These catalysts should be active in the interaction with NO and have properties to oxidize NO to NO2, which is more favored to NOx conversion under oxidizing conditions, such as the technique of the selective catalytic reduction of NOx, especially with some N-containing as agents like ammonia or urea (NH3-SCR) [6]. Unfortunately, all these catalysts exhibit a lot of disadvantages. For example commonly used commercial catalysts, e.g. V2O5–WO3/MnO3 or V2O5–WO3/TiO2, at high temperatures oxidize SO2 to SO3, which can give sulfuric acid and, as a consequence, result in corrosion and deposition of solid by-products downstream from the reactor, where the temperature is lower. Another disadvantage of V2O5 is its toxicity. The metal oxides, such as unsupported MnOx or MnOx/Al2O3 have low deNOx selectivity to N2 and produce great amounts of the N2O. The noble metals, on the other hand, are expensive, suffer due to deactivation by poisoning with the sulfur compounds and have weak thermal stability, which leads to active phase aggregation. Other catalysts like zeolites have low hydrothermal stability [6–11]. A very promising alternative for the above mentioned catalysts is ceria–zirconia mixed oxide (CZ), which has attracted much attention of the researchers working on the NOx reduction in the last 20 years. The main advantages of CZ are good stability and unique redox properties. The labile oxygen vacancies and bulk oxygen species with relatively high mobility are easily formed during the redox shift between Ce3+ and Ce4+ under oxidizing and reducing conditions, respectively. The pure CeO2 is rarely used as a catalyst or support for NOx reduction because it has poor thermal stability. The addition of ZrO2 improves not only thermal stability but also the oxygen storage capacity and redox properties of ceria, upgrading the same its catalytic activity at lower temperatures. Furthermore, the addition of noble or transition metals (or their oxides) has a promotional effect on the redox properties and oxygen storage capacity of CZ [5, 12–14]. The catalytic activity of ceria–zirconia mixed oxides depends on the Ce/Zr ratio. Among a few CexZr(1-x)O2 supports, the Ce0.62Zr0.38O2 has been found to be the most active [15], and such support has been chosen as a support for the copper catalysts used in our studies.
The aim of our work is to prepare and characterize catalysts, which can be potentially used for NOx removal via NH3-SCR. The ceria–zirconia supported copper catalyst (CuO/Ce0.62Zr0.38O2) was synthesized with different copper loadings and characterized using the BET method and powder X-ray diffraction (XRD). The temperature programmed reduction of the catalysts with hydrogen (H2-TPR) was conducted in order to evaluate the variation of their redox properties. Acidic properties have been examined by the NH3 adsorption. The catalysts have also been investigated for their ability to oxidize NO to NO2. According to the literature, NO2 particles are preferentially reduced when the reaction is carried out in the presence of a reductant such as ammonia. For this purpose, the experiments of temperature programmed desorption of NO (NO-TPD) and NO oxidation to NO2 were conducted. Additionally, for better understanding the structure and evolutions of species adsorbed on the catalyst surface, both experiments, i.e. NO-TPD and NO oxidation, were carried out in the DRIFT cell in controlled atmosphere.
Experimental
Catalysts preparation
The ceria–zirconia supported copper catalysts were prepared by the incipient wetness impregnation of the commercial Ce0.62Zr0.38O2 (CZ), provided by Rhodia Electronics & Catalysts, with aqueous solutions of copper nitrate. After impregnation, the samples were dried at 120 °C for 12 h and then calcined at 550 °C for 2. The loading of copper was 2, 4 and 10 wt%. The obtained catalysts CuO/Ce0.62Zr0.38O2 were denoted as Cu(x)/CZ, where x was the loading of Cu.
Characterization of the catalysts
The specific surface area (Ssp) of the support and studied catalysts was determined using the BET method. The Ssp was measured by nitrogen adsorption at 77.35 K on the ASAP 2010 apparatus. Prior to each measurement, the samples were degassed at 105 °C until the stable vacuum of 10−3 Pa was reached. The chemical compositions of the studied catalysts were obtained by ICP.
The X-ray diffraction (XRD) measurements were performed on the Siemens D5005 apparatus (Bruker-AXS) with CuKα radiation, operating at 30 kV and 50 mA. The data were collected in the 2θ range of 10°–90°. The phase identification was made using the JCPDS cards.
The experiments of temperature-programmed reduction (H2-TPR) were performed using 50 mg samples of the support and studied catalysts. The samples were reduced in hydrogen 5 % (v/v) H2/Ar (flow 20 ml min−1) from room temperature to 900 °C with the heating rate of 7.5 °C min−1. The consumption of hydrogen was monitored by the thermal conductive detector.
The experiments of temperature-programmed desorption of NO (NO-TPD) were performed using a BELCAT-M apparatus. First, NO was adsorbed at room temperature in the presence of oxygen (500 ppm NO and 5 % (v/v) O2/Ar) for 60 min. The NO-TPD experiment was carried out in flowing 5 % (v/v) O2/Ar from RT to 500 °C with a heating rate of 4 °C min−1. In all experiments, the total flow rate was 250 ml min−1 and the gas hourly space velocity was 10.000 h−1. The concentration of NO and NO2 gas at the detector outlet was analyzed using NDIR detector.
The evolution of surface species on Cu/CZ catalysts during their interaction with 500 ppm of NO + 5 % vol. O2/Ar at programmed temperature was studied. The in situ FTIR spectra of adsorbed species were recorded in the range of 4000–500 cm−1 using a BIORAD FTS 165 spectrometer equipped with the high temperature environmental DRIFT cell. The total gas flow was 20 ml min−1. In the first step, the samples were pretreated in flowing Ar at 400 °C for 0.5 h in order to remove water and other impurities from the catalysts surface. Then, the sample was cooled in Ar to the room temperature. The background spectra were collected from RT to 400 °C in flowing Ar. The spectra of catalyst surface in NO+O2/Ar were also collected from RT to 400 °C after 20 min of isotherm in given temperature. The same procedure was performed during interaction catalyst surface with 500 ppm NH3/Ar.
The DRIFTS spectra during the temperature programmed desorption of NO (NO-TPD) were recorded as follows: first, the reaction mixture (500 ppm of NO + 5 vol. % of O2/Ar) was adsorbed on the catalyst surface at 20 °C for 60 min; then, the surface was purged with Ar for 30 min; and finally, the temperature was increased from RT up to 400 °C in flowing Ar. The spectra of catalyst surface were collected after 10 min of remaining at each temperature.
Before the experiments, each sample was calcined in air for 2 h at 500 °C with the rate of heating 10 °C min−1 and cooled down to room temperature.
Results and discussion
Catalyst composition and specific surface area
The chemical composition and SSA of the support and obtained catalysts are presented in Table 1. It is observed that the specific surface area decreases with increasing copper content, which can be due to the sintering of ceria–zirconia , as a consequence of impregnation process and further calcination.
X-ray diffraction (XRD)
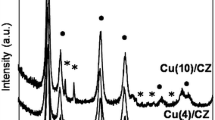
The ceria–zirconia solid solutions CexZr1-xO2 exist in the three stable phases: monoclinic, tetragonal, cubic and two metastable phases [16]. Fig. 1 presents the XRD pattern of the studied support and catalysts. All the diffractograms show the main reflexes typical to a fluorite cubic structure. The reflections corresponding to (111), (200), (220), (311), (222), (400) and (420) planes are observed [17]. In addition, no segregation of the individual phases such as CeO2 or ZrO2 was detected, which can be due to the incorporation the Zr4+ cations into the ceria lattice. The radius of Zr is 0.084 nm and is smaller than the radius of Ce (0.097 nm). Such incorporation leads to formation of homogenous solid solutions [16, 18]. The XRD pattern of Cu(2)/CZ and Cu(4)/CZ (Fig. 1b and c) do not show any typical reflections of the CuO phase. It can be caused by the detection limits of this technique or due to high dispersion of the metal oxide phase. When the CuO content was increased to 10 wt% (Fig. 1d), the CuO crystallites formed agglomerates and the reflexes corresponding to (200), (111), (013), (022) and (222) planes of CuO were observed [19].

XRD patterns of the support CZ (a) and catalysts: Cu(2)/CZ (b), Cu(4)/CZ (c) and Cu(10)/CZ (d)
Temperature programmed reduction (H2-TPR)
Fig. 2 shows the H2-TPR profiles of the support and studied catalysts. The positions of the reduction peaks (T max) and values of H2 consumption are presented in Table 2. The H2-TPR profile of pure ceria–zirconia revealed the presence of two peaks at a temperature of 377 and 557 °C. These peaks suggest that the reduction of CZ occurs in two steps. At lower temperature, the surface ceria is reduced, whereas the reduction of bulk ceria occurs at higher temperature [12, 20]. The reduction of ceria Ce4+ occurs according to the following equation Eq. (1) [21]:

H2-TPR profiles of the support and Cu(2)/CZ, Cu(4)/CZ, Cu(10)/CZ catalysts
The oxygen anions are eliminated as water molecules and oxygen vacancies are formed, which promotes the mobility of oxygen from the bulk to the surface, according to Eq. 2, where Vo it is oxygen vacancy [22] The H2 consumption arises only from the reduction of Ce4+ to Ce3+ because Zr4+ is the non-reducible cation [23].
Fig. 2 shows the H2-TPR profiles for the Cu/CZ catalysts. Each TPR profile can be divided into two reduction areas. The first area from 100 to 180 °C can be ascribed to the reduction of surface ceria or highly dispersed CuO. Moreover, the presence of copper oxide changes the shape of the support reduction curve and shifts the reduction into lower temperature. The second reduction area from 200 to 220 °C can be attributed to the reduction of copper oxide [21, 24], which occurs according to Eq. 3:
Additionally, the reduction of copper oxide requires the presence of a starting point such as an oxygen vacancy on the surface, which can be formed by the reduction of surface ceria [20]. Table 2 presents temperature of the reduction peaks (T max) and values of H2 consumption. The H2 consumption is the smallest for the support, and addition of CuO leads to an increase in hydrogen consumption from Cu(2)/CZ to Cu(10)/CZ. Increasing redox properties with the increasing loading of Cu can be explained not only by the reducibility of CuO but also by the strong interaction between the copper and support. Copper is responsible for the increase of oxygen mobility. Therefore, the support is reduced at lower temperature and value of hydrogen consumption for catalysts is higher [23, 25].
Temperature programmed desorption of NO (NO-TPD) and in situ DRIFTS
The temperature programmed desorption of NO (NO-TPD) has been used for characterizing the support and catalysts from the point of view of NO activation on their surfaces. The NO-TPD experiments have been complemented by the in situ DRIFTS, which is extremely useful method for studying the binding states of adsorbed species. Fig. 3 and Table 3 show the NO-TPD curves and amounts of adsorbed and desorbed NOx species for the studied catalysts. As shown in Fig. 3a, two peaks of NO2 desorption occur for both the support and copper catalysts. The low temperature peaks may be ascribed to desorption of the weakly physisorbed NO2 species. The high temperature desorption peaks may be attributed to the strongly adsorbed NO2 species [26–28]. Analogously, two peaks of desorption are observed for the NO curve (Fig. 3b). It can be observed that the introduction of CuO to ceria–zirconia shifts the desorption peaks both for NO and NO2 into lower temperature. The addition of the metal phase (CuO) shift a peaks of desorption to the lower temperature.

NO2 (a) and NO (b) evolution during NO-TPD carried out in the presence of 500 ppm NO, 5 % O2 on CZ and Cu(2)/CZ, Cu(4)/CZ, Cu(10)/CZ catalysts
The amount of the adsorbed NOx species per 1 g of catalyst increase with the addition of active phase and is the highest for Cu(4)/CZ. The lowest amount adsorbed NOx species is for Cu(10)/CZ, which can be explained by the blocking of active sites in the CuO phase and, thus, the difficulty in the adsorption of NOx species. Another reason can be explained by the formation of agglomerates by CuO, while for the catalyst with 2 and 4 % amount of copper CuO can be well dispersed and the active sites are more available for NOx adsorption. It is interesting that the amount of adsorbed NO and desorbed NOx species on the studied catalysts is different. These phenomena can be explained by strong chemical adsorption of NO on the catalyst surface. Therefore, completely desorption of adsorbed NO species is difficult to observing in studied region of the temperature.
The results of in situ DRIFTS spectra Cu(10)/CZ during NO by O2 at the catalyst surface at the temperatures ranging from RT to 400 °C are shown in Fig 4. According to Hadjiivanov [29] and other authors [30–32], nitrogen oxides can be adsorbed on the catalyst surface forming nitrite (NO2 −) or nitrate (NO3 −) anions. When NO2 − and NO3 − ions are coordinated with cations by one or two of its oxygen atoms, the corresponding species are called nitrito and nitrato species, respectively. Nitrite (NO2 −) and nitrate (NO3 −) species can be monodentate, bidentate or bridging. Generally, the presence of bands at 1470–1375 cm−1 (υ N=O) and 1206–1065 cm−1 (υ N–O) corresponds to monodentate nitrito species. Consequently, the bands at 1378 and 1453 cm−1, which are observed in Fig. 4, can be attributed to nitrite species. These bands are observed up to 200 °C, and they decrease at higher temperatures. Simultaneously, absorptions at 1528, 1609 cm−1 (υ3 (N–O)), indicate formation of monodentate and bridging nitrato species. The bands at 1239 and 1196 cm−1 can be attributed to the nitrate or nitrite species. Hence, nitrites and nitrates are formed at low temperatures.

DRIFTS spectra of Cu(10)/CZ surface during adsorption of the 500 ppm NO, 5 % O2
The surface oxygen interacts with NO from the gas phase to yield nitrite, which is related to electron transfer to the surface of catalyst [30]:
The formation of some NO3 − species at lower temperature may involve the surface reaction between the nitrite species with oxygen from the surface, Eq. 5, or as the result of the disproportionation of nitrite species, Eq. 6:
The electrons in Eqs. 4 and 5 participate in the reduction of the active phase. When the temperature is increased, nitrites are transformed to nitrates. Consequently, nitrates can be decomposed to NO2, Eq. 7, or can react with NO from the gas phase, which results in nitrite and NO2 species, Eq. 8.
At high temperature, the NO2 species are decomposed to NO according to thermodynamic equilibrium, Eq. 9.
This can explain the presence of bands coming from NO2 − and NO3 − surface species (Figs. 4, 5) and the presence of NO and NO2 on NO-TPD profile (Fig. 3) [30, 32].

DRIFT spectra of Cu(10)/CZ surface after adsorption of the 500 ppm NO, 5 % O2
The above results were confirmed by DRIFTS studies of Cu/CZ catalysts during NO-TPD. The NO-TPD experiments were preceded by the adsorption of NO+O2/Ar. The DRIFTS spectra during NO-TPD (Fig. 5) revealed the presence of bands at 1475 cm−1, which can be assigned to stretching vibrations of N–O bond in NO2 − structure. The intensity of these bands increases with temperature up to 200 °C and then decreases. Simultaneously, three bands at 1534, 1237 and 1033 cm−1 are shown. Their intensities are low below 200 °C but they increase significantly above this temperature. The DRIFTS spectra of Cu(10)/CZ during NO-TPD have shown that nitrites transform into nitrates when temperature increases. Moreover, the highest rate of the formation of surface nitrates is observed from 200 to 300 °C. It is due to thermodynamic limitation, because simultaneously nitrates decompose into nitrites above 300 °C. Hence, further increase of absorptions up to 400 °C for bands at 1534, 1237 cm−1 is practically not observed.
DRITS of NH3 adsorption
Because ammonia is considered as an active reductant in process of NOx reduction, it is important to know in which way ammonia interacts with the catalyst surface. It is well known that one of the methods for determined adsorption ammonia and surface acidity is DRIFT [31].
The DRIFT spectra of adsorbed species on the Cu/CZ surface resulting from ammonia adsorption at different temperatures are shown in Fig. 6. According to many authors, adsorption of ammonia can proceed at the Brønsted or Lewis acid sites, forming NH4 + or adsorbed NH3 [33, 34]. When ammonia is adsorbed on the Brønsted acid sites, the bands in the 1480–1395 cm−1 region of absorption can be observed [2, 10, 34]. Thus, the bands at 1459 and 1395 cm−1 for Cu(10)/CZ are attributed to these ammonia species. The absorption bands assigned to the ammonia adsorbed on the Lewis acid sites are observed at 1211 and 1647 cm−1 [2, 10, 33]. For the Cu(10)/CZ catalyst, the bands at 1647 cm−1 disappear at 200 °C, whereas absorptions band at 1211 cm−1 increase up to 400 °C. During the temperature programmed surface reaction of the Cu(10)/CZ catalyst with ammonia, the bands corresponding to NH3 adsorbed on the Lewis and Brønsted acid strength decrease. Therefore, new bands at 1528 cm−1 are observed and they can be attributed to the NH2 species. We proposed the formation of NH2 species on the catalysts surface as the results of H or 2H abstraction from coordinated NH3 or NH4 + on the catalyst surface, respectively, Eqs. 10 and 11 [2, 10, 33].

DRIFT spectra of Cu(y10)/CZ surface during adsorption of the 500 ppm NH3
The electrons forming in the above reactions are transfer to the surface of catalyst. The absorption assigned to the coordinated NH3 species are observed at 1300 cm−1 and other bands, appearing at 1070–1020 cm−1 region and can be assigned to the adsorption of ammonia on the surface oxygen atoms via hydrogen bonding [33].
The surface acidity was confirmed by the adsorption of ammonia both on the Lewis and Brønsted acid sites. Moreover, during temperature programmed surface reaction these acid sites have been disappeared and the band new absorptions of NH2 species are formed.
Conclusions
The CuO/CZ catalysts with different amounts of copper oxide were synthesized. The structural, redox, acidic and adsorption properties were investigated. Surface acidity was studied by DRIFT spectra of the adsorbed NH3 species. The ammonia was adsorbed on the Lewis and Brønsted acid sites and it was transformed into the NH2 species when temperature was increased. From the NO-TPD results, it was calculated that the amount of adsorbed NO species increased after addition of the copper. It was the highest for Cu(4)/CZ. The CuO could be well dispersed in the case of Cu(4)/CZ catalyst. For this metal loading, the active sites are more available for NO adsorption than for Cu(10)/CZ catalyst, in which CuO can form agglomerates, and consequently it is difficult to adsorb NO species. The presented work has shown that the ceria–zirconia supported copper catalysts are able to oxidize NO to NO2 even at low temperature. It can be explained by the strong oxidizing properties of the surface oxygen of ceria–zirconia. From the DRIFT spectra, it was observed that NO can adsorbed on the catalysts surface to form nitrate and nitrite species. Nitrites are formed by electron transfer from NO to the catalyst and they are transformed to nitrate species via oxygen transfer promoted letter in presence of NO2. The nitrite species are subsequently transformed into nitrates when temperature increases up to 300 °C. The rate of surface nitrate formation also increases with temperature. Above 300 °C, the absorption bands of the nitrates disappear because of their decomposition to nitrites which are due to thermodynamic limitations. The results of DRIFT spectroscopy were consistent with the results of NO-TPD experiments. By the observation of the functional groups evolution, several surface reactions have been proposed.
References
Zhu J, Thomas A (2009) Perovskite-type mixed oxides as catalytic material for NO removal. Appl Catal B 92:225–233
Si Z, Weng D, Wu X, Li J, Li G (2010) Structure, acidity and activity of CuOx/WOx–ZrO2 catalyst for selective catalytic reduction of NO by NH3. J Catal 271:43–51
Casapu M, Bernhard A, Peitz D, Mehring M, Elsener M, Kröcher O (2011) A Niobia-Ceria based multi-purpose catalyst for selective catalytic reduction of NOx urea hydrolysis and soot oxidation in diesel exhaust. Appl Catal B 103:79–84
Iwamoto S, Takahashi R, Inoue M (2007) Direct decomposition of nitric oxide over Ba catalyst supported on CeO2-based mixed oxides. Appl Catal B 70:146–160
Gómez-García MA, Pitchon V, Kiennemann A (2005) Pollution by nitrogen oxides: an approach to NO x abatement by using sorbing catalytic materials. Environ Intern 31:445–467
Boyanoa A, Lázaroa MJ, Cristiani C, Maldonado-Hodarc FJ, Forzatti P, Molinera R (2009) A comparative study of V2O5/AC and V2O5/Al2O3 catalysts for the selective catalytic reduction of NO by NH3. Chem Eng J 149:173–182
Liu F, He H, Ding Y, Zhang Ch (2009) Effect of manganese substitution on the structure and activity of iron titanate catalyst for the selective catalytic reduction of NO with NH3. Appl Catal B 93:194–204
Xu W, Yu Y, Zhang Ch, He H (2008) Selective catalytic reduction of NO by NH3 over a Ce/TiO2 catalyst. Catal Comm 9:1453–1457
Irfan MF, Goo JH, Kim SD (2008) Co3O4 based catalysts for NO oxidation and NOx reduction in fast SCR process. Appl Catal B 78:267–274
Liu F, He H, Zhang Ch, Shen W (2011) Mechanism of the selective catalytic reduction of NOx with NH3 over environmental-friendly iron titanate catalyst. Catal Today 175:18–25
Pietrogiacomi D, Magliano A, Sannino D, Campa MC, Ciambelli P, Indovina V (2005) In situ sulphated CuOx/ZrO2 and CuOx/sulphated-ZrO2 as catalysts for the reduction of NOx with NH3 in the presence of excess O2. Appl Catal B 60:83–92
Damyanova S, Pawelec B, Arishtirova K, Martinez Huerta MV, Fierro JLG (2008) Study of the surface and redox properties of ceria–zirconia oxides. Appl Catal A 337:86–96
Liu L, Yu Q, Zhu J, Wana H, Sun K, Liu B, Zhu H, Gao F, Dong L, Chen Y (2010) Effect of MnOx modification on the activity and adsorption of CuO/Ce0,67Zr0,33 catalyst for NO reduction. J Colloid Interface Sci 349:246–255
Adamowska M, Krzton A, Najbar M, Camra J, Djega-Mariadassou G, Da Costa P (2009) Ceria–zirconia-supported rhodium catalyst for NO x reduction from coal combustion flue gases. Appl Catal B 90:353–544
Adamowska M, Muller S, Da Costa P, Krzton A, Burg P (2007) Correlation between the surface properties and deNOx activity of ceria–zirconia catalysts. Appl Catal B 74:278–289
Adamski A, Tabor E, Gil B, Sojka Z (2007) Interaction of NO and NO2 with the surface of Ce x Zr1-x O2 solid solutions—influence of the phase composition. Catal Today 119:114–119
JCPDS 01-078-0694
Atribak I, Guillén-Hurtado N, Bueno-López A, García-García A (2010) Influence of the physico-chemical properties of CeO2–ZrO2 mixed oxides on the catalytic oxidation of NO to NO2. Appl Surf Sci 256:7706–7712
JCPDS 04-009-2287
Ayastuy JL, Gurbani A, Gonzales-Marcus MP, Gutierrez-Ortiz MA (2012) Selective CO oxidation in H2 streams on CuO/CexZr1−xO2 catalysts: correlation between activity and low temperature reducibility. Int J Hydrogen Energy 37:1993–2006
Trovarelli A, De Leitenburg C, Dolcetti G, Lorca JL (1995) CO2 methanation under transient and steady-state conditions over Rh/CeO2 and CeO2-promoted Rh/SiO2: the role of surface and bulk ceria. J Catal 151:111–124
Gutierez-Ortiz JI, De Rivas B, López-Fonsea R, Gonzàles-Velasco JR (2005) Characterization of the catalytic properties of ceria–zirconia mixed oxides by temperature programmed techniques. J Thermal 80:225–228
Li G, Wang Q, Zhao B, Zhao R (2010) Modification for Ce0,67Zr0,33O2 mixed oxides by coprecipitated/impregnated Co: effect on the surface and catalytic behaviour of Pd only three-way catalyst. J Mol Catal A 326:69–74
Zhou R, Yu T, Jiang X, Chen F, Zheng X (1999) Temperature programmed reduction and temperature-programmed desorption studies of CuO/ZrO2 catalysts. Appl Surf Sci 148:263–270
Teng M, Lou L, Yang X (2009) Synthesis of mesoporous Ce1−xZrxO2 (x = 0.2–0.5) and catalytic properties of CuO based catalysts. Microporous Mesoporous Mater 119:158–164
Wu X, Lin F, Xu H, Weng D (2010) Effects of adsorbed and gaseous NOx species on catalytic oxidation of diesel soot with MnOx–CeO2 mixed oxides. Appl Catal B 95:101–109
Wang Y, Zhu A, Zhang Y, Au CT, Yang X, Shi C (2008) Catalytic reduction of NO by CO over NiO/CeO2 catalyst in stoichiometric NO/CO and NO/CO/O2 reaction. Appl Catal B 81:141–149
Li WB, Yang XF, Chen LF, Wang JA (2009) Adsorption/desorption of NOx on MnO2/ZrO2 oxides prepared in reverse microemulsions. Catal Today 148:75–80
Hadjiivanov KI (2000) Catal Rev Sci Eng 42:71
Adamowska M, Krzton A, Najbar M, Da Costa P, Djega-Mariadassou G (2008) DRIFT study of the interaction of NO and O2 with the surface of Ce0.62Zr0.38O2 as deNO x catalyst. Catal Today 137:288–291
Little LH (1966) Infrared spectra of adsorbed species. Academic Press, London
Azambre B, Zenboury L, Delacroix F, Weber JV (2008) Adsorption of NO and NO2 on ceria–zirconia of composition Ce0.69Zr0.31O2: a DRIFTS study. Catal Today 137:278–282
Davydov A (2003) Molecular spectroscopy of oxide catalyst surface. Wiley, England
Casapu M, Krocher O, Mehring M, Nachtegaal M, Borca C, Harfouche M, Grolimund D (2010) Characterization of Nb-containing MnOx–CeO2 catalyst for low temperature selective catalytic reduction of NO with NH3. J Phys Chem C 114:9791–9801
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Radlik, M., Adamowska, M., Łamacz, A. et al. Study of the surface evolution of nitrogen species on CuO/CeZrO2 catalysts. Reac Kinet Mech Cat 109, 43–56 (2013). https://doi.org/10.1007/s11144-013-0552-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-013-0552-7