
Abstract
The kinetics of the 1,6-intramolecular hydrogen migration in the alkyl radical reaction class has been studied using the reaction class transition state theory (RC-TST) combined with the linear energy relationship (LER) and the barrier height grouping (BHG) approach. The RC-TST/LER, where only reaction energy is needed, and RC-TST/BHG, where no other information is needed, are found to be promising methods for predicting rate constants for any reaction in the 1,6-intramolecular H migration in alkyl radicals reaction class. Direct comparison with available experimental data indicates that the RC-TST/LER, where only reaction energy is needed, can predict rate constants for any reaction in this reaction class with satisfactory accuracy.
Similar content being viewed by others


Introduction
Being the initial product from the radical attack on the alkanes, alkyl radicals are important intermediates in the decomposition of organic fuels. It has long been known that the alkyl radical reactions play an important role in the combustion and atmospheric degradation pathways of traditional fuels as well as a wide variety of other chemical processes [1–3]. The long chain hydrocarbons are the most important components of both traditional fuels and biofuels. To optimize its fuel potential, a detailed knowledge of the long chain alkyl radicals kinetics is needed. Their chemistry is largely governed by mutually competitive unimolecular reactions, namely intramolecular H atom migration and direct decomposition (beta scission) of the C–C bond. For this reason, there has been considerable and growing interest in investigating both of these reaction families for a long time [4–33].
In spite of the significant progress made, relatively little information is still known for the kinetics of the reactions mentioned above. Especially, little is known about the macroscopic features of reactions of long chain radicals. This is because direct measurements of the rate constants of decomposition are difficult due to the competition between the multichannel reactions with short lived and highly reactive intermediates. A series of shock tube results with a quoted uncertainty of less than a factor of 2, for both n-alkyl and branched alkyl radicals, were reported by Tsang and co-workers [11–13]. The authors presented high pressure limits of rate constants for all possible H migrations and C–C bond beta scissions of the n-hexyl, n-octyl and 4-methyl-1-pentyl radicals. There are also a number of theoretical studies on the activation energies, transition state (TS) geometries and rate constants of several small alkyl radical decompositions. Davis et al. [4] investigated all possible terminal hydrogen migration pathways for the selected n-alkyl radicals. The observable differences for axial and equatorial forms of the radicals as well as the effect of the abstraction site location relative to the terminal carbons were noticed. The same studies were also performed for branched alkanes [5], where the effect the methyl group has on the reaction enthalpy, activation energy, and rate coefficients of the 1,2 through 1,7 H-migration reactions was also assessed. Recently, the same authors extended their studies to cyclic radicals [6]. Wang et al. [10] theoretically studied the competitive unimolecular decomposition and isomerization of the 1-hexyl radical. The authors successfully reproduced experimental data reported by Tsang et al. [12]. The internal H isomerization channel was found as dominant over the beta scissions in the low temperature (250–900 K) region. A similar conclusion was derived by Curran et al. [3, 34] in their series of proposed hydrocarbon combustion mechanisms. However, the alkyl radical decomposition channel was found to be more important in the high temperature region (T >850 K). In general, β-scission has been well accepted as the dominant decomposition path for alkyl radicals. Thus, accurate kinetic data for both β-scission and intramolecular H migration of a wide range of alkyl radicals are of importance.
Since it is not merely arduous, but simply impossible to study all elementary reactions over the whole P–T parameter space experimentally, theoretical methods that enable the extrapolation of the kinetics data to the full range of parameters encountered in combustion studies are necessary. Accurate methodologies were developed in the past. The reaction class transition state theory (RC-TST) [35] extrapolates a known rate constant of selected reaction (called reference reaction) from a given class to that of any arbitrary reaction in the same class using correlations, which are constructed under the TST framework. The key idea of this application is that reactions in the same class have the same reactive moiety, whose chemical bonding changes during the course of the reaction, and thus, they are expected to have similarities in their potential energy surfaces along the reaction pathways/valleys (see Fig. 1 in the ref [36]). The group additivity (GA) approach is mainly based on the fact that reaction rates depend primarily on the thermodynamic properties of the involved species, and that thermal properties can be predicted on the basis of the assumption of GA. To derive the rate constants, this method takes advantage of the similarity in the reactive moiety of reactions in the same class to define a “supergroup” for the TSs. The GA approach was successfully applied by Sumathi et al. [37, 38], Sabbe et al. [24–26] and Wang et al. [39]. Another method, where rate rules are derived from a systematic investigation of sets of reactions within a given reaction class using electronic structure calculations performed at the CBS-QB3 level of theory, was reported by Villano et al. [40]. The survey of the different rate estimation rules was recently reported by Carstensen et al. [41], the applicability of the particular approaches is also assessed. Recently, the new methodology wherein the fuel surrogate is defined in terms of pseudo species including the functional groups contained in the actual fuel was reported and successfully applied to model of the real combustion systems [42].
An application of the RC-TST method for intramolecular H migrations in alkyl radicals was reported in the literature [28]. A variant of the RC-TST method, easily applicable to the automated reaction mechanism generator (ARMG)—software, called RC-TST/linear energy relationship (LER) was also reported in the literature [35] and applied to a number of different reaction classes [27, 29, 43, 44]. In particular, RC-TST/LER correlations were derived for the 1,4 [27] and 1,5 [29] H migrations. For the long chain alkyl radicals, 1,6-H migrations are also of importance [4, 5, 11, 14, 28]. For that reason, the RC-TST/LER correlations for the 1,6-H migration in alkyl radicals reaction family are reported in this study.
Methodology
Reaction class transition state theory (RC-TST)
Since the details of the RC-TST method have been presented elsewhere [35], we discuss only its main features here. It is based on the realization that the reactions in the same class have the same reactive moiety; thus the difference between the rate constants of any two reactions is mainly due to differences in the interactions between the reactive moiety and their different substituents. Within the RC-TST framework, the rate constant of an arbitrary reaction (denoted as k a ) is proportional to the rate constant of a reference reaction, k r , by a temperature-dependent function f(T):
One often would choose the reference reaction to be the smallest (i.e. with the smallest reactant molecules) reaction in the class, since their rate constants can be calculated accurately from first principles. The key idea of the RC-TST method is to factor f(T) into different components under the TST framework:
where \( f_{\sigma } \), \( f_{\kappa } \), \( f_{Q} \), \( f_{V} \) and \( f_{HR} \) are the symmetry number, tunneling, partition function, potential energy and hindered rotations (HRs) factors, respectively. These factors are simply the ratios of the corresponding components in the TST expression for the two reactions:
where κ(T) is the transmission coefficient accounting for the quantum mechanical tunneling effects; σ is the reaction symmetry number; Q ≠ and ΦR are the total partition functions (per unit volume) of the TS and reactants, respectively; ΔV ≠ is the classical reaction barrier height; c HR symbolizes the correction to the total partition function due to the HR treatment, T is the temperature in kelvin; k B and h are the Boltzmann and Planck constants. Among these, only the symmetry factor can be easily calculated from the molecular topology of the reactant. Obtaining exact values of four other factors requires structures, energies, and vibrational frequencies of the reactants and TS of the reaction investigated. The potential energy factor can be calculated using the reaction barrier heights of the arbitrary reaction and the reference reaction. The RC-TST/LER method uses the LER similar to the well-known Evans–Polanyi linear free-energy relationship [45] between classical barrier heights and reaction energies of reactions to estimate reaction barriers and determines the pre-exponential factor (relative to a well characterized reference reaction) by performing a cost-effective molecular mechanics or density functional theory (DFT) calculation with statistical analysis. Furthermore, this variant of the RC-TST method uses averaged values of the partition, tunneling and HR factors rather than their exact values defined by Eqs. (4–7). As a consequence, the RC-TST/LER rates constants are estimated using only reaction energy and reactant topology information; no TS and frequency calculation are needed. This feature makes RC-TST/LER method applicable to different ARMG schemes. Alternatively, it is possible to approximate all reactions at the same type of carbon atom site as having the same barrier height, namely the average value. In previous studies [27, 29, 43, 44], we found that for the different reaction classes, average values of the reaction barriers are dependent on the order of the active carbon site; this approximation was referred to as the barrier height grouping (BHG) approximation. It was shown [27, 29, 36] that substitution of an alkyl group will stabilize the radical species thus lowering the barrier height. Thus, one may expect hydrogen migration reactions from tertiary carbon to have lower barrier heights than those from secondary carbon. The same relationship is expected to hold between H-shifts from secondary and primary H abstraction sites.
Computational details
All the electronic structure calculations were carried out using the GAUSSIAN 09 suite of programs [46]. A hybrid non-local DFT, particularly Becke’s half-and-half (BH&H) non-local exchange and Lee–Yang–Parr (LYP) non-local correlation functionals, has previously been found to be sufficiently accurate for predicting the TS properties for different classes of reactions [27, 28, 35, 47]. Note that within the RC-TST framework as discussed above, only the relative barrier heights are needed. Our previous studies have shown that the relative barrier heights for the title reaction family can be accurately predicted by the BH&HLYP method. Geometries of reactants, TSs, and products were thus optimized at the BH&HLYP level of theory with Dunning’s correlation-consistent polarized valence double zeta basis set [3s2p1d/2s1p] denoted as cc-pVDZ, which is sufficient to capture the physical change (i.e. transformation of the reactant into cyclic TS and breaking the TS ring to create product) along the reaction coordinate for this type of reaction. All reported results for stable molecules as well as TSs were obtained for the lowest energy conformer of a given species. Normal mode analysis is performed at each stationary point to ensure its characteristics, i.e. stable structure has zero imaginary vibrational frequency whereas TS structure has one imaginary vibrational frequency, whose mode corresponds to the reaction coordinate of the reaction being considered. Geometry, energy, and frequency information were used to derive the RC-TST factors.
To derive the RC-TST correlation functions TST/Eckart rate constants for all reactions in the representative set were calculated. All kinetic calculations were done using the TheRate [48] program. In these calculations, all molecular rotations were treated classically and vibrations treated quantum mechanically within the harmonic approximation except for the modes corresponding to the internal rotations of the –CH3 groups, which were treated as HRs using the method of Ayala et al. [49, 50]. It is important to point out that both the motion of the internal rotation of the methyl group in the reactive moiety and internal rotors gain and loss in the course of the reaction is already treated explicitly in the rate constants of the reference reaction R1 [28]. Thus, the reaction class factor due to these HRs is a measure of the substituent effects on the rate constant from the hindered rotors relative to that of the reaction R1. To calculate the HR correction factor to the partition function for a certain vibrational mode, the rotating group and the periodicity number of the torsional potential of the vibrational mode must be identified. From the given information together with the geometry of the interested molecule, data needed for calculating the correction factor of HR treatment, e.g. reduced moment of inertia and the periodic potential can be obtained. The correction factor is then calculated using the fitting formula (Eq. 26 in Ref. [49]). A previous study by Kungwan and Truong [47] has shown that the contribution of HRs from alkyl groups larger than –CH3 is relatively small due to the cancellation of the treatment within the RC-TST framework. For this reason, we only consider HR treatment for the –CH3 groups in this study. To do this, periodic torsional potential is needed. We found that the rotational barriers depend mainly on the kind of the carbon atoms to which the methyl group is directly connected. These barriers were found to be equal 3.5, 4.0, 4.4, and 3.7 kcal/mol for the –CH3 groups bonded to the primary, secondary, tertiary, and TS member C atoms. Within any of these three sets, differences in the barriers were found to be negligible, i.e. less than 0.3 kcal/mol. The potential energy curves for these rotations are plotted in Fig. S1 in the Supplementary information.
Thermal rate constants were calculated for the temperature range from 300 to 3,000 K, which is sufficient for many combustion applications.
Results and discussion
In the section below, we describe how the RC-TST/LER factors for the 1,6-H migration in alkyl radicals are derived using the training reaction set, three error analyses are also presented.
As mentioned in the “Introduction” section, the RC-TST/LER correlations for the 1,6-H shift reaction family are needed to provide a complete picture of the internal H migrations in alkyl radicals. The aim of this section is then to obtain all parameters for estimating the rate constants of any intramolecular 1,6-H migration of alkyl radicals. This is done by first deriving analytical correlation expressions for rate constants of the reference reaction with those in a small representative set of the class from explicit direct DFT calculations. The assumption is that these correlation expressions can be extended to all reactions in the class. So far, this assumption has shown to be valid [27, 29, 35, 43, 47]. To develop the RC-TST/LER parameters for the title reaction class, the representative set consists of 18 reactions as shown in the Table 1.
The reference reaction is the 1,6-H migration in the 1-hexyl radical (R1). Of these 18 reactions, 7 represent H shift from a primary C atom (type p), 6 from a secondary carbon (type s) and 5 from a tertiary carbon (type t).
Rate constants of the reference reaction
The first task for applying the RC-TST method to any reaction class is to have rate constants of the reference reaction as accurate as possible. The rate constants can be from either experimental data or first principles calculations. In this study the smallest reaction in the investigated reaction class, called also the principal reaction, namely 1-hexyl → 1-hexyl, is chosen as the reference reaction. Reaction R1 was extensively studied in Ref. [28], its rate constants calculated with the CVT/SCT method for the temperature range of 300–3,000 K were proven to agree well with available experimental data. The CVT/SCT derived rate expression for the reference reaction is as follows [28]:
Reaction class parameters
This section describes how the RC-TST/LER factors were derived using the representative reaction set.
Potential energy factor
To calculate the potential energy factor for the particular reaction its classical barrier is needed. As mentioned previously, the barriers for H abstraction from primary, secondary and tertiary carbon sites are expected to differ. These expectations were confirmed in our DFT calculations, where the title reaction class can be divided to three subclasses, namely H shifts from primary (p), secondary (s) and tertiary (t) carbon atoms. The potential energy factor for each subclass is represented by a different LER. Of the reactions from training set, 7 belong to the p, 6 to the s and 5 to t subclasses. The reaction energies and barrier heights for all representative reactions in the representative set are given explicitly in Table 2. The three derived LERs (one for each subclass) plotted against the reaction energies calculated at the BH&HLYP/cc-pVDZ of theory are shown in Fig. 1a–c. These linear fits were obtained using the least square fitting method and have the following expressions:

Linear energy relationship plot of the barrier heights, \( \Updelta V^{ \ne } \) versus the reaction energies \( \Updelta E \). Both reaction energies and barrier heights were calculated at the BH&HLYP/cc-pVDZ level of theory
Except for the reference reaction R1, the largest absolute deviations of reaction barrier heights between the LERs and the direct DFT BH&HLYP/cc-pVDZ calculations is equal to 0.9 kcal/mol with the medium average deviation (MAD) equal to 0.28 kcal/mol (see Table 2). These deviations are, in fact, smaller than the systematic errors of the computed reaction barriers from full electronic structure calculations (see, for example, Table 1 in Ref. [51]). This is certainly an acceptable level of accuracy for kinetic modeling. Note that in the RC-TST/LER methodology, only the relative barrier height is needed. To compute these relative values, the classical barrier height of the reference reaction R1 calculated at the same level of theory, i.e. BH&HLYP/cc-pVDZ, is needed and has the value of 16.20 kcal/mol [51]. For the BHG approach, the average barrier heights are 23.45, 22.32 and 20.28 kcal/mol with deviations of 1.51, 1.43 and 1.46 kcal/mol for the p, s and t subclasses. These deviations are acceptable in the high temperature region; it may lead to large errors for lower temperatures, however. The key advantage of this approach is that it does not require any additional information to estimate rate constants.
In conclusion, the barrier heights for any reaction in the title reaction class can be obtained by using either the LER or BHG approach. The estimated barrier height is then used to calculate the potential energy factor using Eq. (6). The performance for such estimations on the whole representative reaction set is discussed in the error analyses below.
Reaction symmetry number factor
The reaction symmetry number factors f σ were calculated simply from the ratio of reaction symmetry numbers of the arbitrary and reference reactions using Eq. (3). The symmetry number of an elementary process is equal to the ratio of the total symmetry number of the reactant divided by the total symmetry of the TS [52, 53]. For the H-atom intramolecular migration reaction class, this number is strictly related to the number of H atoms connected to the hydrogen abstraction site: 3 for primary carbons, 2 for secondary, and 1 for tertiary. This value is multiplied by the number of equivalent migration sites in the molecule. In any case, this number can be easily calculated from the molecular topology of the reactant, thus the symmetry number factor can be calculated exactly. The underlying assumption to this practice is that each hydrogen at a given abstraction site has the same kinetic parameters and, thus, the same rate. As pointed out by Davis et al. [5], this may not be the case for some branched radicals with chiral TS. Because there is no easy way to assess magnitude of such effect they are neglected. Direct comparison of the RC-TST results with experiment (see Fig. 5 in Ref. [29]) clearly showed that this approximation is valid for the 1,5-H migrations, its performance for the 1,6-H migrations is evaluated in this study in the same way (see Fig. 4).
Tunneling factor
The tunneling factor f κ is the ratio of the transmission coefficient of reaction R a to that of the reference reaction R r . Due to the cancellation of errors in calculations of the tunneling factors, it was shown that the factor f κ can be reasonably estimated using the one-dimension Eckart method (see Fig. 3 and Tables 2–4 in Ref. [54]). Calculated results for the representative reaction set can then be fitted to an analytical expression. It is known that the tunneling coefficient depends on the barrier height. For the 1,6-H migration in alkyl radicals reaction class, the barrier heights group together into three groups, namely, primary, secondary, and tertiary carbon sites (see “Potential energy factor” section), and it is expected that reactions in the same group have a similar tunneling factor and, thus, the average value can be used for the whole group. Simple expressions for the three tunneling factors for primary, secondary, and tertiary carbon sites are obtained by fitting to the average calculated values are shown in Fig. 2 and were fitted to analytical expressions as given below:

Plots of the tunneling ratio factors \( f_{\kappa } \) as functions of the temperature for the 1,6-hydrogen migration from primary, secondary and tertiary carbon sites
Partition function factor
The partition factor is the product of the translational, rotational, internal rotation, vibrational, and electronic component. The translational and rotational factors are temperature-independent. The main factors that govern the temperature dependence of the f Q factor are the differences in the vibrational frequencies due to the coupling of substituents with the reactive moiety (see Fig. 1 in Ref. [35]), which arises from the vibrational component and internal rotations only. Note that contributions from the HR modes are treated separately, and they are not included in these partition function factors. The temperature dependent partition factor is averaged over all reactions of a given subclass. Averaged partition function factors for the whole class, calculated over the temperature range of 300–3,000 K, is almost constant. For the sake of simplicity, this value can be effectively fitted as:
The average value of partition function factor for the 1,6-H migration in alkyl radicals reaction class differs from unity. As mentioned earlier, the coupling between substituents with the reactive moiety is believed to account for these differences.
HR factor
Individual factors for particular reactions R2–R18, tantamount to the k HO /k HR values for these reactions, are plotted in Fig. S2 and listed in Table S1 of the Supplementary Information. The average value of the HR factor for the 1,6-H migration reaction class, plotted in Fig. 3, is close to unity and is nearly temperature independent. The largest deviation from unity is about 4 %. For simplicity, we assumed that this factor is equal to unity.

Average hindered rotation corrections to the total rate constants for all reactions in the temperature range of 300–3,000 K
Prediction of rate constants
What we have established so far are the necessary parameters—namely potential energy factor, reaction symmetry number factor, tunneling factor and partition function factor— for the application of the RC-TST/LER theory to predict rate constants for any reaction in the 1,6-H shift in alkyl radicals reaction class. The procedure for calculating rate constants of an arbitrary reaction in this class is: (i) calculate the potential energy factor using Eq. (6) with the \( \Updelta V_{r}^{ \ne } \) value of 16.2 kcal/mol. The reaction barrier height can be obtained using the LER approach by employing Eqs. (9a–9c) for BH&HLYP/cc-pVDZ reaction energies or by the BHG approach; (ii) determine the symmetry number factor from Eq. (3); (iii) evaluate the partition function factor using Eq. (11); and (iv) the rate constants of the arbitrary reaction can be calculated by taking the product of the reference reaction rate constant given by Eq. (8) with the reaction class factors above. Table 3 summarizes the RC-TST/LER parameters for this reaction class. If the BHG barrier heights and average values for other factors are used, the rate constants are denoted by RC-TST/BHG. The RC-TST/BHG rate constants for any reactions belonging to this class can be estimated without any further calculations as:
The appropriate symmetry factors are included in the rate constant expressions above. The correction for the number of equivalent reaction paths depends on specific reaction and thus must be included explicitly.
Error analyses
As mentioned earlier, only limited amount of the experimental data is available for intramolecular H shift in alkyl radicals. Experimental data are available for the reactions R6 and R7 from the training set [11]. The experiments were performed in a single pulse shock tube at temperatures in the 850–1,000 K range. High-pressure rate constants have been derived over 700–1,900 K range with an uncertainty factor of less than 2. The value of a given rate constant, k, could lie between k m /f and k m *f, where k m is the reported value and f is an uncertainty factor. A comparison of these data with results reported in this study is presented in Fig. 4a, b. In this figure, RC-TST labeled curves represent results obtained within the full RC-TST methodology, using Eqs. (3–7) to extrapolate high pressure limits of rate constants calculated for the reference reaction R1 to those presented in Fig. 4. To obtain these results, the knowledge of the TS is necessary, whereas only reaction energy is needed in the RC-TST/LER approximation. As can be seen from Fig. 4, the agreement between the RC-TST and RC-TST/LER results and the experimentally derived data [11] is quite satisfactory. This is not always the case for the RC-TST/BHG approach, assuming one averaged barrier height per specific reaction site, however. This approach may introduce a noticeable error (see Fig. 4b), especially in the low temperature region.

Arrhenius plots of the calculated and experimental rate constants for the reactions: a 3-octyl → 1-octyl, b 1-octyl → 3-octyl. Experimental data are taken from Ref. [11]
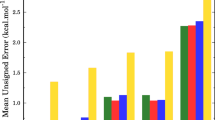
The systematic errors introduced by the LER and BHG approaches are discussed in detail in the next error analysis, which compares RC-TST/LER and RC-TST/BHG results with those from explicit calculations. As mentioned in our previous studies [35, 36], the RC-TST methodology can be thought of as a procedure for extrapolating rate constants of the reference reaction to those of any reaction in the same class. Comparisons between the calculated rate constants for a small number of reactions using both the RC-TST/LER or RC-TST/BHG and the full RC-TST methods provide additional information on the accuracy of the LER and BHG approximations. The results for this error analysis for 17 representative reactions, (i.e., the comparisons between the RC-TST/LER and full RC-TST methods) are shown in Fig. 5a, wherein the relative deviation defined by (|kRC-TST − k RC-TST/LER|/k RC-TST) as a percent versus the temperature for all reactions in the representative set, R2–R18 is plotted. For temperatures larger than 1,000 K and all the reactions in this set, the unsigned relative errors are within 60 %. In the low temperature region, four reactions have errors larger than 100 %. So, in general, it can be concluded that RC-TST/LER can estimate thermal rate constants for reactions in this class within 60 % when compared to those calculated explicitly using the full RC-TST method. For other cases, the maximum error is, except for reaction R3, less than 150 %, which may be still an acceptable level of accuracy for reaction engineering purposes. A similar analysis is presented for the RC-TST/BHG approach as shown in Fig. 5b. As expected, RC-TST/BHG has the larger errors in the low temperature region, specifically 4 from 17 reactions have the maximum error significantly exceeds 100 %. In the low temperature region, 6 reactions exhibit very large errors, which may significantly affect the accuracy of the RC-TST/BHG method. Note that the convenience of readily usable rate expressions for any reaction in the class may still off-set the lower accuracy of the BHG approach compared to that of the LER.

Relative absolute deviations as functions of the temperature between rate constants calculated from explicit full RC-TST calculations for all selected reactions and: a From the RC-TST/LER method where BH&HLYP reaction energies were used for the LER. b From the RCT-TST/BHG method
To demonstrate the reliability of the correlations, further validation is needed to verify that the 18 reaction representative set selected for developing the RC-TST/LER parameters is sufficient to represent this reaction class. We calculated the relative deviation defined by (|k RC-TST − k RC-TST/LER|/k RC-TST) for 10 additional reactions, denoted as R1A–R10A, not included in the training set. These reactions are listed in Table 4. The results are plotted in Fig. 6. Of the reactions R1A–R10A, those with n-cetyl (C16H33) radicals (reactions R3A–R10A), which are currently used to model diesel fuel surrogates[34], are of particular interest to the combustion community. As can be seen from Fig. 6, kinetic data for these migrations in long chain alkyls can be accurately estimated by the RC-TST/LER method. The errors are within the same range as for reactions from the training set (R2–R18), thus proving the validity of the RC-TST/LER approximation.

Relative absolute deviations as functions of the temperature between rate constants calculated from explicit full RC-TST calculations for the reactions R1A–R10A and from the RC-TST/LER method where BH&HLYP reaction energies were used for the LER
Finally, an analysis on the systematic errors in different factors in the RC-TST/LER methods was performed. The total error is affected by the errors in the approximations in the potential energy factor, tunneling factor and partition function factor introduced in the method. The deviations/errors between the approximated and exact factors within the TST framework are calculated at each temperature for every reaction in the representative set and then averaged over the whole class. For the LER approach, the error in the potential energy factor comes from the use of an LER expression; that of the tunneling factor, from using Eqs. (10a–c) and that of the partition function factor, from using Eq. (11). Absolute errors averaged over all 17 reactions, R2 ~ R18 as functions of the temperature are plotted in Fig. 7. Of the factors, the HR and partition function ratios factor show the lowest temperature dependence for the whole temperature range. For T >500 K, the tunneling factor introduced the smallest error of less than 2 %, while the error introduced by the partition function factor is less than 15 %. For T <500 K, the error of the BHG potential energy factor is noticeable and exceeds 200 %. This affects the low temperature behavior of the total BHG factor, which reaches almost 200 % for T = 500 K. This observation confirms the previous conclusions that the BHG approach may be risky for T < 1,000 K. Thus, the LER approach gives noticeably less error in the potential energy factor than the BHG approximation. For the temperature range T >1,000 K, all the errors, except those resulted from the BHG approximation, are almost constant. Except for the BHG approach, the total systematic errors due to the use of simple analytical expressions for different reaction class factors are less than 70 % for the temperature range 300–3,000 K. This value increases for the BHG approach reaching 500 % for T = 300 K (not visible in Fig. 7), thus the RC-TST/BHG is rather not recommended for T <1,000 K. The failure of the BHG method for the title (and also for the 1,5 internal H migrations [29]) reaction classes is due to the inadequacy of basic assumption underlying this approximation, namely simple dependence of the barrier height on the type of the H abstraction site. As reported by Davis et al. [4, 5], the reaction barrier depends not only on the abstraction site, but also on a number of the other issues such as destination site, location of the abstraction site relative to terminal carbon atom, the presence of methyl substituents in the TS ring in the case of branched alkyl radicals and conformation.

Averaged absolute errors of the total relative rate factors f(T) (Eq. 2) and its components, namely the tunneling (\( f_{K} \)), partition function (\( f_{Q} \)), potential energy (\( f_{V} \)) and hindered rotation(f HR ) factors as functions of temperature
Summary of approximations in the RC-TST method
By generalizing from the small reference reaction to larger homologues and, consequently, enabling the obtaining of any rates constants within a given reaction class with accuracy comparable to high level methods but at a fraction of the cost, the RS-TST method provides an effective way to derive considerable benefits from expensive electronic structure calculations. However, the user should be aware of the approximations used. In particular, these are as follows:
-
The accuracy of RC-TST rate constants depends on the accuracy of the rate constants of the reference reaction. Thus, each improvement in the rate measurement/calculation methodology results in considerable enhancement of the predicted rates.
-
The recrossing effect is taken into account only in part, corresponding to those in the reference reaction. The “additional” recrossing trajectories, not included in the reference reaction rate constants, are neglected.
-
Although absolute transmission coefficients for hydrogen abstraction reactions often require multidimensional tunneling methods to account for the large corner-cutting effects, it was shown [35, 54] that, because of cancellation of errors, the tunneling factor f κ can be accurately predicted using the 1-D Eckart method, as it is done in the RC-TST/LER approach.
-
The RC-TST method does not take into account conformational aspects, which may be important in some of the reactions within the family, especially for branched alkyl radicals.
-
Since the number of HR modes may be different for reactants and TSs, the f HR factor may not properly capture the differences of the HRs partition functions of the reference reaction and some other processes within the family, thus affecting the resulting rates constants. However, as shown in Fig. 3, this error is not significant for the 1,6-H transfer reaction class; for other families of errors resulting from using the harmonic approximations are also mostly canceled. Since the entropy changes (internal rotors gain and lost) during the course of the reaction are already included in the principal reaction (see Ref. [28]), they are believed not to affect the results. Similarly, it is also believed that internal rotations of the –CH2 groups are mutually cancelled.
-
In the RC-TST/LER approximation, the barrier height for any reaction within the family is calculated with the LER. Although, as shown in Table 2, the error associated with this approximation is not large, it may affect the low temperature value of the potential energy factor f v and, consequently, the RC-TST/LER rate constants.
Conclusions
The application of the reaction class TS theory combined with the linear energy relationship and the barrier height grouping approach to the prediction of thermal rate constants for hydrogen 1,6-intramolecular hydrogen migration reaction class was carried out. The rate constants for the reference reaction, 1-hexyl → 1-hexyl, were obtained by the CVT/SCT method in the temperature range 300–3,000 K. The RC-TST/LER, where only reaction energy is needed, was found to be a promising method for predicting rate constants for any reaction in this reaction class. The error analyses indicate that this method can predict rate constants within a factor of 2 compared to explicit rate calculations. The RC-TST/BHG method, where no other information is needed, can also be employed for the quick estimation of reaction rates in the high temperature region.
References
Simmie J (2003) Prog Energy Comb Sci 29:599–634
Zádor J, Taatjes CA, Fernandes RX (2011) Prog Energy Comb Sci 37:371–421
Curran HJ, Gaffuri P, Pitz WJ, Westbrook CK (1998) Combust Flame 114: 149–177
Davis AC, Francisco JS (2011) J Phys Chem A 115:2966–2977
Davis AC, Francisco JS (2011) J Am Chem Soc 133:19110–19124
Davis AC, Tangprasertchai N, Francisco JS (2012) Chem Eur J 18:11296–11305
Dobe STB, Reti F, Marta P (1987) Int J Chem Kinet 19:895–921
Matheu DM, Green WH, Grenda JM (2003) Int J Chem Kinet 35:95–119
Xu X, Papajak E, Zheng J, Truhlar DG (2012) Phys Chem Chem Phys 14:4204–4216
Wang F, Cao DB, Liu G, Ren J, Li YW (2010) Theor Chem Acc 126:87–98
Tsang W, McGivern WS, Manion JA (2009) Proc Combust Inst 32:131–138
Tsang W, Walker JA, Manion JA (2007) Proc Combust Inst 31:141–148
McGivern WS, Awan IA, Tsang W, Manion JA (2008) J Phys Chem A 112:6908–6917
Sirjean B, Dames E, Wang H, Tsang W (2012) J Phys Chem A 116:319–332
Yamauchi N, Miyoshi A, Kosaka K, Koshi M, Matsui H (1999) J Phys Chem A 103:2723–2733
Viskolcz B, Lendvay G, Körtvélyesi T, Seres L (1996) J Am Chem Soc 118:3006–3009
Viskolcz B, Lendvay G, Seres L (1997) J Phys Chem A 101:7119–7127
Lendvay G, Viskolcz B (1998) J Phys Chem A 102:10777–10786
Caralp F, Devolder P, Fittschen C, Gomez N, Hippler H, Mérau R, Rayez MT, Striebel F, Viskolcz B (1999) Phys Chem Chem Phys 1:2935
Hippler H, Viskolcz B (2000) Phys Chem Chem Phys 2:3591–3596
Hippler H, Striebel F, Viskolcz B (2001) Phys Chem Chem Phys 3:2450–2458
Jitariu LC, Jones LD, Robertson SH, Pilling MJ, Hiller IHJ (2003) J Phys Chem A 107:8607–8617
Peng S, An R, Wu Z, Li Y (2012) Reac Kinet Mech Cat 107:105–113
Sabbe MK, Vandeputte AG, Reyniers MF, Van Speybroeck V, Waroquier M, Marin GB (2007) J Phys Chem A 111:11771–11786
Sabbe MK, Reyniers MF, Waroquier M, Marin GB (2010) ChemPhysChem 11:195–210
Vandeputte AG, Sabbe MK, Reyniers M-F, Marin GB (2012) Phys Chem Chem Phys 14:12773–12793
Bankiewicz B, Huynh LK, Ratkiewicz A, Truong TN (2009) J Phys Chem A 113:1564–1573
Ratkiewicz A, Bankiewicz B, Truong TN (2010) Phys Chem Chem Phys 12:10988–10995
Ratkiewicz A, Bankiewicz B (2012) J Phys Chem A 116:242–254
Alecu IM, Truhlar DG (2011) J Phys Chem A 115:14599–14611
Zheng J, Truhlar DG (2010) Phys Chem Chem Phys 12:7782–7793
Curran HJ (2006) Int J Chem Kinet 38:250–275
Engel PS, Gudimetla VB, Gancheff JS, Denis PA (2012) J Phys Chem A 116:8345–8351
Curran HJ, Gaffuri P, Pitz WJ, Westbrook CK (2002) Combust Flame 129:253–280
Zhang S, Truong TN (2003) J Phys Chem A 107:1138–1147
Truong TN (2000) J Chem Phys 113:4957–4964
Sumathi R, Carstensen HH, Green WH Jr (2001) J Phys Chem A 105:8969–8984
Sumathi R, Green WH (2002) Theor Chem Acc 108:187–213
Wang HX, Wang BY, Zhang JL, Li ZR, Li XY (2011) Chem J Chin Univ 32:1123–1128
Villano SM, Huynh LK, Carstensen H-H, Dean AM (2011) J Phys Chem A 115:13425–13442
Carstensen HH, Dean AM (2009) J Phys Chem A 113:367–380
Mehl M, Pitz WJ, Sarathy SM, Westbrook CK (2012) Int J Chem Kinet 44:257–276
Ratkiewicz A, Truong TN (2010) Int J Chem Kinet 42:414–429
Ratkiewicz A, Bieniewska J, Truong TN (2011) Int J Chem Kinet 43:78–98
Polanyi JC (1972) Acc Chem Res 5:161–168
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng, G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, Revision B.01. Wallingford, CT
Kungwan N, Truong TN (2005) J Phys Chem A 109:7742–7750
Duncan WT, Bell RL, Truong TN (1998) J Comput Chem 19:1039–1052
Ayala PY, Schlegel HB (1998) J Chem Phys 108:2314–2325
Zheng J, Yu T, Papajak E, Alecu IM, Mielke SL, Truhlar DG (2011) Phys Chem Chem Phys 13:10885–10907
Ratkiewicz A (2011) Phys Chem Chem Phys 13:15037–15046
Pollak E, Pechukas P (1978) J Am Chem Soc 100:2984–2991
Fernandez-Ramos A, Ellingson BA, Garret BC, Truhlar DG (2007) Rev Comput Chem 23:125–232
Truong TN, Duncan WT, Tirtowidjojo M (1999) Phys Chem Chem Phys 1:1061–1065
Acknowledgments
Author thanks the computational center of the University of Warsaw (ICM) for providing access to the supercomputer resources and the GAUSSIAN 09 program (Grant G33-03).
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ratkiewicz, A. Kinetics of 1,6-hydrogen migration in alkyl radical reaction class. Reac Kinet Mech Cat 108, 545–564 (2013). https://doi.org/10.1007/s11144-012-0518-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-012-0518-1