
Abstract
Objectives
To assess the ability of a previously developed hybrid physiology-based pharmacokinetic-pharmacodynamic (PBPKPD) model in rats to predict the dopamine D2 receptor occupancy (D2RO) in human striatum following administration of antipsychotic drugs.
Methods
A hybrid PBPKPD model, previously developed using information on plasma concentrations, brain exposure and D2RO in rats, was used as the basis for the prediction of D2RO in human. The rat pharmacokinetic and brain physiology parameters were substituted with human population pharmacokinetic parameters and human physiological information. To predict the passive transport across the human blood–brain barrier, apparent permeability values were scaled based on rat and human brain endothelial surface area. Active efflux clearance in brain was scaled from rat to human using both human brain endothelial surface area and MDR1 expression. Binding constants at the D2 receptor were scaled based on the differences between in vitro and in vivo systems of the same species. The predictive power of this physiology-based approach was determined by comparing the D2RO predictions with the observed human D2RO of six antipsychotics at clinically relevant doses.
Results
Predicted human D2RO was in good agreement with clinically observed D2RO for five antipsychotics. Models using in vitro information predicted human D2RO well for most of the compounds evaluated in this analysis. However, human D2RO was under-predicted for haloperidol.
Conclusions
The rat hybrid PBPKPD model structure, integrated with in vitro information and human pharmacokinetic and physiological information, constitutes a scientific basis to predict the time course of D2RO in man.
Similar content being viewed by others

Introduction
In schizophrenia drug therapy and research, dopamine D2 receptor occupancy (D2RO) is often used as a target biomarker to quantify the relationship between efficacy and side effects (1). Several studies suggest that blockade of 65 to 80% of D2 receptors is the key to antipsychotic efficacy for both conventional neuroleptics and novel antipsychotics (2–4). D2RO higher than 80% increases the risk of adverse effects such as extra pyramidal symptoms (5). Thus, D2RO has a central role in antipsychotic drug discovery, drug development and therapy. Target occupancy is important both in early drug discovery, where accurate knowledge of the degree of occupancy could help to determine the suitability of a drug candidate for further development, and later in the drug development process, when target site occupancy measurements can guide dose selection (6). D2RO is clinically measured using positron emission tomography (PET) or single-photon emission computed tomography (SPECT) methodology, which are both expensive and time-consuming. Tools to predict clinical D2RO in preclinical drug discovery phases are therefore valuable. It is well known that current antipsychotics also activate or antagonize other targets in the central nervous system. For example, risperidone has a higher affinity for serotonin (5-HT2A) receptors than for D2 receptors (5). Extensions of this tool to other receptors would therefore increase the value of the current translation framework.
Recently, we have reported physiology-based pharmacokinetic-pharmacodynamic (PBPKPD) models to characterize the time course of D2RO and 5-HT2A receptor occupancy (5-HT2ARO) in rats (7,8). The mechanistic and physiological basis of these models should potentially allow the prediction of human PKPD properties using physiological parameters and prior information from in vitro and in vivo preclinical studies (9). The present investigation aimed to determine how these models can be used for translating receptor occupancy from rat to humans.
Development of a translation tool to predict human D2RO (based on PKPD models) involves scaling information from rat to human. This involves accounting for drug distribution to the brain and the drug’s ability to bind to striatal D2 receptors. Drug distribution to the brain is not only characterized by passive diffusion but also by active efflux transporters present at the luminal surface of the blood–brain barrier (BBB). Similarities in the in vitro permeability values determined by various types of experiments provide a basis to integrate and scale information on passive drug transport to the brain from in vitro to in vivo or from one species to the other (10). However, differential expression and limited homology of drug transporters involved in active drug transport across the BBB leads to challenges when scaling active transport related information from one species to another (11). Notwithstanding divergent reports on the species independence of drug-specific parameters, integration of in vitro parameters with physiologically based PKPD modeling would increase the potential of successfully translating effects from preclinical species to humans (12).
Hence, the objective of this work was to explore different approaches to predict human striatal D2RO using a generic translational PBPKPD model structure, which allows integration and scaling of information from preclinical in vitro or in vivo data to the human situation. Different approaches were compared to determine the minimal amount of information required for this translational work. A previously developed extended model structure (8) was also used to predict human 5-HT2ARO based on these approaches.
Methods
Data
This work was performed within the framework of the Dutch Top Institute Pharma project: Mechanism-based PK–PD modeling (http://www.tipharma.com). This mechanism-based PKPD modeling platform involves leading pharmaceutical companies worldwide, and academic institutes from the Netherlands. Three pharmaceutical companies who are the members of this mechanism-based PK–PD modeling platform, namely, Janssen Research and Development - Belgium, Merck Sharp and Dohme - The Netherlands and Pfizer Worldwide Research and Development – USA, provided human plasma concentration data for haloperidol (HAL), risperidone (RIS) and paliperidone (PAL, in extended release formulation) and helped with collection of human D2 and 5-HT2A receptor occupancy data. Clozapine (CLZ), HAL, olanzapine (OLZ), RIS, extended release formulation of PAL and quetiapine (QTP) were used in this study as model antipsychotic drugs. The observed human D2RO for these antipsychotics were taken from the literature (2,13–24). For risperidone (RIS), human D2RO was provided from the pharmaceutical companies who are involved in this project.
Population pharmacokinetic (PK) parameters for CLZ, OLZ, QTP and RIS were obtained from literature (1,25,26). However, no population PK models have been reported in literature for HAL and PAL. So, for these compounds in-house population PK models were developed. The population PK model for haloperidol was developed on the basis of data from 7 studies, comprised of 122 individuals [healthy volunteers (n = 20) and schizophrenic patients (n = 102)] and 515 plasma concentrations obtained across a wide dose range of 1 to 60 mg/day administered either as single or multiple doses. The population PK model for paliperidone-extended release was developed on the basis of data from 3 studies, comprised of 870 individuals and 4169 plasma concentrations obtained across a wide dose range of 3 to 15 mg/day administered as an OROS® once daily formulation.
Physiology-Based PKPD Model Structure
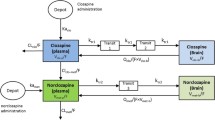
A PBPKPD model was previously developed and evaluated for its usefulness in describing the time course of brain concentration and D2RO in rats (7). This model contains expressions to describe the kinetics in brain-vascular, brain-extravascular, striatum-free and striatum-bound compartments (Fig. 1). Following administration, drug is transported from the plasma compartment to the brain-vascular compartment; this process is assumed to be determined only by the cerebral blood flow. Only the unbound drug in this vascular compartment crosses the BBB and is transported into the brain-extravascular compartment, which is governed by the brain-extravascular clearance (CLbev). Furthermore, drug is transported from the brain-extravascular compartment to the striatal-free compartment. The brain-extravascular and striatum-free compartments were assumed to be equilibrating rapidly. In striatum-bound compartment, drug can reversibly bind to the dopamine receptor complex (Fig. 1). The receptor association and dissociation processes were described using kon as the receptor association rate constant (nM−1h−1), koff as the receptor dissociation rate constant (h−1) and D2 receptor density in striatum (nM).

A schematic representation of the PBPKPD model. The model incorporates different processes to explain the time course of D2RO. The brain pharmacokinetics describes the processes involved in the transport of drug from plasma to brain, and the striatum compartment explains the drug binding to receptors through the binding constants.
In addition to the previously developed rat PBPKPD model structure, an active efflux clearance (CLeff) component between brain-extravascular and brain-vascular compartments was included in this predictive model to reflect the active drug transport from brain, when appropriate (i.e., for RIS and PAL). Additionally, this rat PBPKPD model structure was extended to account for binding of RIS and PAL to the 5-HT2A receptor (8). This extended model included two additional compartments (cortex-free and cortex-bound). Binding to 5-HT2A receptors was described using association and dissociation constants and receptor density values specific for 5-HT2A receptors. This extended model structure was used to predict both D2 and 5-HT2A receptor occupancy in humans in this simulation study.
Human D2RO predictions
The rat PBPKPD model structure (Fig. 1) was used to predict the D2RO versus time profile of antipsychotics in humans by substituting all parameters of the rat model by their human analogues, according to the following methods:
-
(1)
Human population PK parameters estimated using total plasma concentrations were obtained either from models developed in-house or from published literature (Table I).
-
(2)
Physiological parameters, such as blood flow to the brain and the brain volumes were obtained from literature (Table II).
-
(3)
Passive permeability transport across the BBB was scaled from in vitro or in vivo rat to in vivo human based on the assumption that permeability for passive diffusion per cm2 of brain endothelial surface area is identical the between different systems.
-
(4)
Active efflux transport for PAL and RIS was scaled using pertinent information on Pgp protein expression in the different systems as a scaling factor.
-
(5)
Receptor binding was either derived from in vitro Ki values or in vivo Kd values corrected for differences between the in vitro and the in vivo system of the same species.
-
(6)
Experimentally determined values of the fraction unbound in human plasma and in rat brain were obtained from the literature. Unbound fraction in human brain is assumed to be equal to the unbound fraction in rat brain.
Different approaches were used to obtain human parameters and they were detailed as Approaches A-C in this section.
-
Approach A was based only on human in vitro information (in vitro apparent permeability, efflux ratio (ER), in vitro Ki and koff).
-
Approach B was based on the in vivo parameters (rCLbev, rCLeff, Kdrat, koff-rat) obtained from the rat PBPKPD model.
-
Approach C was aimed at using minimal information to get the best predictions of human D2RO by integrating approaches A and B.
All the parameters values used for these simulations are presented in Table III.
Approach A: Human D2RO predictions based on in vitro information
Passive drug transport at the BBB
Experimentally determined in vitro apparent permeability (Papp) values were used to predict passive transport of antipsychotics across the BBB. Specifically, values of the v permeability across multidrug resistance Madin-Darby canine kidney (MDR1-MDCK) type II cell monolayers were obtained from Summerfield et al. (32). Permeability determined while attenuating transporters denotes the ability of the molecule to traverse membranes by passive means (41). These Papp values were translated to a meaningful parameter of human brain extra-vascular clearance (hCLbev) across the BBB by taking the product of Papp and human brain endothelial surface area of 20 m2 (27).
Passive and active drug transport at the BBB
For the antipsychotics PAL and RIS that are known to have both active and passive transport across the BBB, the drug transport component was derived from Papp and in vitro efflux ratio (ER) determinations. ER is commonly used as an indicator of active drug transport in CNS drug discovery. ER is calculated as the ratio of effective permeability for a drug from the basal side to the apical side to that in the opposite direction (37). ER was used to calculate the human active efflux clearance (hCLeff) of RIS and PAL. The parameters describing hCLbev and hCLeff accounting for active transport were derived based on Kwon et al. (42), underlying derivations are presented in Appendix 1.
hCLbev and hCLeff represent the passive permeability and active transport clearance across the BBB in humans, respectively. Papp represents the in vitro apparent permeability and HBSA represents human brain endothelial surface area of 20 m2..
Receptor binding parameters
In vitro Kihuman values were used as the parameter Kd (equilibrium constant). If available, in vitro or ex vivo experimentally determined koff values were used in these simulations (Table III). If experimental koff values were not available, then calculated koff values based on Ki and koff correlation of different antipsychotics were used, as reported previously (5).
Approach B: Human D2RO predictions based on in vivo information
The appropriateness of using the available rat hybrid PBPKPD model to determine in vivo parameters for human D2RO predictions was assessed. Binding constants (Kon and Koff) and clearances (rCLbev and rCLeff) were obtained using PBPKPD models developed by us previously (7,8). This in vivo model based information was only available for OLZ, PAL and RIS.
Passive drug transport to the brain
Calculated Papp (Pappcalc) values were derived as the ratio between PBPKPD model-estimated in vivo rCLbev and rat brain endothelial surface area (150 cm2/g) and then normalized for an average rat brain weight of 2 g/250 g of rat (32). The product of Pappcalc and the human brain endothelial surface area (20 m2) was used as the hCLbev - the passive transport clearance across the human BBB.
Active drug transport from the brain
Human active efflux clearance was predicted based on the PBPKPD model-estimated rCLeff and mdr1a protein expression in micro vessels in mouse. Mdr1a expression values for rats were not available; hence, mdr1a expression in rat brain was assumed to be equal to that of mouse. Mouse mdr1a expression was documented as 14.1 fmol/μg of protein, and MDR1 expression in human brain was documented as 6.06 fmol/μg of protein (11). The following Eq. (Eq 3) describes the assumed relationship that was used to derive the hCLeff.
Receptor binding parameters
Model estimated in vivo kon-rat and koff-rat values were used as in vivo human kon and koff assuming that these drug-specific parameters do not require any scaling between species (9). For OLZ, model estimated in vivo Kdrat and koff-rat values were taken from our previous publication (7). For RIS and PAL, in vivo binding constants were obtained from the extended model structure where the time courses for D2 and 5-HT2A receptor binding were modeled together (8). These parameters are shown in Table IV.
Approach C: Human D2RO predictions integrating in vitro and in vivo information
Passive and active drug transport to the brain
Scaling and calculation used in Approach A were also applied in the integrated Approach C.
Receptor binding parameters
In vivo Kdhuman parameters were corrected for the differences between in vitro and in vivo scenarios by normalizing model estimated in vivo Kdrat and in vitro Ki values for rat and human, as shown in Eq. (Eq 4).
In vivo koff-human values were assumed to be equal to in vitro or ex vivo experimentally determined koff values.
Human 5-HT2ARO predictions
The objective of this exercise was to check the utility of the extended model structure to predict human D2RO and 5-HT2ARO. In vitro Kihuman and Kirat for PAL were 0.250 nM (43). In vitro Kihuman and Kirat for RIS were 0.160 and 0.210 nM, respectively (43,44). We were unable to find koff value for 5-HT2A binding from any in vitro source and so, the in vivo based Approach B and the integrated Approach C were applied for these predictions.
Human D2 receptor occupancy simulations
For each approach (A-C), 1000 human D2RO- time course curves were simulated at clinically relevant doses, administered orally. Differential equations (Appendix 2) explaining the pharmacokinetics and pharmacodynamics of antipsychotics were used in these simulations as implemented in R (Version 3.1.0) using the deSolve package (45). Inter-individual variability (IIV) in the population pharmacokinetic parameters was accounted for in these simulations. The predictive power of this translational approach was determined by comparing these simulations with observed human D2RO. In clinical trials, human D2RO information were collected after repeated dosing and at steady state conditions. Hence, time course of D2RO was simulated and results at steady state (achieved within 2 or 3 weeks of repeated drug dosing) were compared with the observed steady state RO.
Predictions of human RO were performed for CLZ (500 mg/day), HAL (2 mg/day), OLZ (10 mg/day), PAL (9 mg/day), QTP (750 mg/day) and RIS (4 mg/day) dose levels and compared graphically with observed D2RO. Additionally, for OLZ, RIS and PAL box plots of prediction errors were made to compare the applicability of the different approaches and their predictive power. For this purpose, D2RO or both D2RO and 5-HT2ARO predictions were made for a drug treatment of 3 weeks, at a single time point (12 h after the last dose of OLZ, RIS and 2 h after the last dose of PAL). These predictions were then compared with the median of the actual observations. The selection of dose and time points was based on the availability of data.
% Prediction Error (PE) was calculated as follows:
The median D2RO observed experimentally for OLZ, PAL and RIS were 54.9, 77.4, 75.8%, respectively. The 5-HT2ARO observed for RIS was 100% (46).
Results
Human D2RO predictions
Approach A
This approach predicted the time course of human D2RO well for five of the antipsychotics, but not for HAL, as depicted in Fig. 2. The percentage prediction bias of these predictions is depicted for OLZ, PAL and RIS in Table V and Fig. 3.

Observed and predicted steady-state D2 receptor occupancy in humans after oral administration of antipsychotics at clinically relevant doses. Simulations were performed using the rat PBPKPD model structure integrated with in vitro apparent permeability, efflux ratio and in vitro binding information (Approach A). Depicted are the observed D2RO (dots) and the shaded area represent the 95% prediction limits of the simulated D2RO. The medians of the simulated D2RO are represented as a solid line.

Box plots representing the % prediction error (PE) for different approaches where in vitro, in vivo and integrated in vitro and in vivo information were used to predict the human D2RO. Letters A, B and C denotes the three different approaches, Approach A, Approach B and Approach C, respectively.
Approach B
The predictive model based on in vivo model estimated parameters under-predicted the human D2RO for OLZ, RIS and PAL (Table V, Fig. 3).
Approach C
The predictive model using the “corrected” in vivo Kdhuman estimates predicted human D2RO for OLZ are at the best when compared to the other approaches (Fig. 3). For PAL and RIS, the D2RO was slightly over-predicted and the prediction bias was large in comparison to Approach A (Figs. 3 and 4) and Table V.

Observed and predicted steady-state D2 receptor occupancy (D2RO) in humans after oral administration of risperidone or paliperidone at clinically relevant doses. Depicted are the observed D2RO (dots) and the shaded area represent the 95% prediction limits of the simulated D2RO. The medians of the simulated D2RO are represented as a solid line. Panel (a) and Panel (b) represent the human D2RO predictions for paliperidone achieved by approaches A and C, respectively. Panel (c) and Panel (d) represent the human D2RO predictions for risperidone based on approaches A and C, respectively.
Human 5-HT2ARO predictions
The extended predictive model using the “corrected” in vivo Kdhuman estimates predicted the human 5-HT2ARO for risperidone better than approach B. Human D2RO predictions were similar to that of the model which only accounts for the D2 receptor binding, although the prediction bias for Approach B is lower (Table V, Fig. 5).

Box plots represent the % prediction error (PE) for human D2RO and 5-HT2ARO predictions of risperidone at 4 mg/day dose. % PE for human D2RO predictions obtained by both D2 model structure (D2MS) and an extended model structure including 5-HT2ARO (5-HT2AMS) are compared in the left hand panel. Right hand panel represents the human 5-HT2ARO predictions of risperidone obtained from the extended model structure using approach B and C.
Discussion
This study aimed to utilize the recently proposed rat PBPKPD model structure (7,8) in a translational framework to scale pharmacokinetic and pharmacodynamic information from rats to human. The objective of this work was also to determine the minimal information required to be included in this translational framework to predict the D2RO during the early drug discovery phase, while taking into account distribution to the brain and receptor binding.
In this simulation study, three approaches (A, B and C) were used to predict D2RO in human. Approach A uses information based on in vitro studies, Approach B uses information obtained from in vivo studies based on rats and Approach C integrates both in vitro and in vivo based information. Due to limitation of in vivo based information, Approach B and Approach C were applied only for olanzapine, risperidone and paliperidone. The following part discusses the implementation of these approaches to translate permeation of drugs to brain and their binding to D2 receptors.
Translation of passive drug transport at the BBB
It is well known that tight junctions and active efflux transporters present at the luminal surface of the BBB are involved in the distribution of any substance to the brain. Hence, hCLbev and hCLeff were included in this model structure to explain passive permeability across the BBB and active efflux processes in a mechanistic manner.
In vitro effective permeability of compounds with various characteristics across human primary brain endothelial cells was comparable to those obtained with bovine and rat capillary endothelial cells (10). So, a simplistic way of translation of passive permeability across the BBB between species is by accounting for the differences in system-specific brain endothelial surface area and utilize the in vitro effective permeability information without any scaling between species. In Approach A, passive permeability of the compounds across the BBB was calculated as the product of human brain endothelial surface area and Papp values obtained from in vitro MDR1-MDCK.
Approach B used the passive permeability of compounds across the BBB under in vivo conditions. This was achieved by estimating model parameter explaining the passive transport of drugs across the BBB by fitting a PBPKPD model to plasma and whole brain concentration data obtained from rats. Pappcalc (permeability across the BBB in vivo) values were derived from PBPKPD model-estimated in vivo rCLbev and rat brain endothelial surface area.
The hCLbev values calculated based on in vitro Papp or Pappcalc (calculated based on rCLbev) values were different (Table III) and this could direct towards a theory on different efficiency of drug transport in in vitro and in vivo systems. However, it should be noted that the PBPKPD model-based estimate rCLbev (and thereby Pappcalc) was based on whole brain concentration and therefore this data do not differentiate the influence of the cerebrospinal fluid barrier and other microenvironment conditions on the transport of drug at the BBB. In addition, Pappcalc was derived using reported rat brain endothelial surface area, which ranges from 100 to 240 cm2 per g brain tissue (47,48). These underlying assumptions and lack of information limited the application of Approach B. Notwithstanding this limitation, deriving a relationship between model-estimated rCLbev (and thereby Pappcalc) and in vitro Papp based on more compounds might help to improve the translation approach. However, the limited number of compounds used in our PKPD analysis did not allow us to elicit such a relationship. So, in the absence of such a relationship, it seems to be more appropriate to base the scaling of in vitro Papp values to hCLbev on human brain endothelial surface area only. Hence, while integrating in vitro and in vivo information (i.e., Approach C), the calculated hCLbev was based only on in vitro Papp value and human brain endothelial surface area.
Translation of active drug transport at the BBB
Predicting active efflux clearance at the human BBB using in vitro information was challenging and complicated. In this simulation study, in vitro ER was used to account for the active drug transport out of the brain. In Approach A, in vitro ER was used to calculate the drug transport across the BBB by means of active transport. Derivation based on Kwon et al. (42) explains the transport of compound across membranes under in vitro conditions using Papp and ER and by using this derivation and HBSA, the parameters hCLbev and hCLeff were calculated for human conditions. This translation approach is simple and based only on information from in vitro. Hence, this will be a useful tool to predict brain transport in human brain environment at early stages of drug development. Nevertheless, it should be noted that this translation approach accounts only for differences in brain endothelial surface area between species and excludes differences in transporter expression levels. Extension of this approach accounting for those differences on microenvironment levels between different species would help to improve predictions of drug transport at BBB.
Subsequently, active efflux transport was also scaled to the human conditions based on the estimates obtained from the rat PBPKPD model parameters and expression of mdr1a protein in both species (i.e., Approach B). As described earlier, this PBPKPD model-based estimate of rCLbev (and thereby Pappcalc) and rCLeff were based on whole brain concentration and therefore had no ability to differentiate microenvironment conditions at the BBB. In addition, due to non-availability of the appropriate mdr1a expression data in rats, it was assumed that it is equal to that of mouse. Additionally, the active transport was included as a linear process rather than non-linear as usually described for in vitro systems, since the free concentrations of drug at the BBB (in rats) were much lower than the concentrations used in vitro, and remain most likely below the Km (concentration require for the half -maximal transport) for the transporter, which makes this assumption acceptable. A proper evaluation of these assumptions would help to substantiate the claims, however due to lack of human brain drug exposure information these assumptions are seldom evaluated. Nevertheless, this work provides a framework to account for the active drug transport in humans. It is noteworthy that both these approaches (Approach A and B) are plausible because of their mechanistic basis.
Translation of receptor binding properties
Danhof et al. (49) proposed that the values of drug-specific parameters such as target affinity are likely to be identical between species and individuals. This would imply that the binding rate constants estimated in rats could be used to extrapolate the pharmacodynamics from rat to human. Hence, it is appropriate to include in vitro or in vivo binding constants in this model structure to predict human D2RO. This predictive model structure was obtained from a preclinical system where the drug binding to D2 receptors was explained by accounting for the association and dissociation rates of antipsychotics. The model estimated in vivo Kdrat values for OLZ were close to in vitro Kirat values (rat cloned D2L system – 17 nM), but in vitro Kihuman values (human cloned D2L system- 5.1 nM) were different from both these values of the rat system. Additionally, PAL and RIS in vitro Kirat values (most commonly reported as 2 nM) are different from the model estimated in vivo Kdrat values (0.364 nM). The human D2RO predictions for OLZ, RIS and PAL were not consistent with the observed human D2RO data when model estimated in vivo Kdrat was used as the in vivo Kdhuman parameter in the predictive model. This challenges the general belief that drug-specific parameters like Kd can be used across species without any scaling. However, this difference between the in vivo and in vitro scenario for the same species could arise from the assumptions used in both in vitro calculations and model estimations. Additionally, radio-ligand selection and disturbances in assumed equilibrium conditions in in vitro and in vivo systems could lead to biased or inappropriate Ki calculations (50).
Extension of predictive model
It has been demonstrated that the binding to both D2 and 5-HT2A receptors (extended model) was essential to explain the relationship between drug exposure and receptor occupancy in the preclinical system with good precision (8). The model estimates of in vivo Kdrat for RIS are influenced by the brain distribution kinetics and it was elucidated that the brain-to-plasma ratio (in rats) is not constant for RIS, suggesting an influence of specific binding to receptors on the brain-kinetics (8). Hence, an extended model structure was used to predict both D2 and 5-HT2A receptor occupancy in humans. The extended structure predicted 5-HT2ARO well. Surprisingly, the D2RO predictions achieved by using these two different model structures (D2 alone versus D2 + 5-HT2A) remained close to each other; and this extension, which was essential for model fitting in a preclinical system, did not significantly improve the human D2RO predictions (atleast for Approach C). Nevertheless, this extended model structure underscores the ability of this model framework to be flexible and extendable to other receptor types.
Our objective was also to study the minimal information required to predict human D2RO. In general, human D2RO was predicted well for all compounds except haloperidol when only in vitro information (Approach A) was used in the simulations. This demonstrates the ability of this model structure to predict human D2RO with minimal in vitro information.
In this simulation study, the time course of plasma concentrations was obtained from available population pharmacokinetic parameters. It is also possible to predict these pharmacokinetic parameters based on in silico and in vitro information using commercially available tools, like Simcyp (Simcyp Ltd., Sheffield, UK). So, the requirement of population pharmacokinetic parameters obtained from clinical studies is not essential.
For HAL, D2RO predictions were lower than the observations. This may be related to the high ratio of unbound concentrations of HAL in brain and plasma, which is close to 4 in rats (51). This high brain to plasma ratio may indicate a unique active influx transport to the brain. In addition, it has been documented that the metabolism of HAL involves a conversion of HAL to reduced haloperidol, and back-conversion of reduced haloperidol to HAL (52) in the brain of guinea pigs. Accounting for this metabolism and/or active influx transport may help to improve predictions. Extending this predictive model structure to include such complexity is practically possible, if sufficient information about each related process is available beforehand.
Applications of this predictive tool are not limited to only predicting D2RO in early drug discovery but also in selecting appropriate first in human doses based on pharmacodynamics. It is not anticipated that predictive tools will completely replace the need for experiments, though it is plausible that this tool can help to design more informative and more efficient clinical studies.
Conclusion
A general translational framework was developed which is based on a mechanism-based approach and accounts for the different processes involved in the transport of drug to the brain. Based on, in vitro information (Papp, ER and Ki), the model was able to predict the human D2RO for drugs distributed to the brain by passive permeability and active transport processes. This model structure with an appropriate extension also predicted the human 5-HT2ARO well.
Abbreviations
- BBB:
-
Blood–brain barrier
- CL:
-
Clearance
- CLbev :
-
Brain extra-vascular clearance (passive permeability clearance across the BBB)
- CLeff :
-
Active efflux clearance across the BBB
- CLZ:
-
Clozapine
- ER:
-
Efflux ratio
- h:
-
Human (prefix)
- HAL:
-
Haloperidol
- HBSA:
-
Human brain endothelial surface area
- Kd:
-
In vivo receptor equilibrium dissociation constant
- Ki:
-
In vitro receptor equilibrium dissociation constant determined in inhibition study
- koff:
-
Receptor dissociation rate constant
- kon:
-
Receptor association rate constant
- MDR1-MDCK:
-
Multidrug resistance Madin-Darby canine kidney
- OLZ:
-
Olanzapine
- PAL:
-
Paliperidone
- Papp:
-
Apparent permeability across the BBB
- PBPKPD:
-
Physiology-based pharmacokinetic-pharmacodynamic model
- PE:
-
Prediction error
- PET:
-
Positron emission tomography
- Pgp:
-
P-glycoprotein
- PK:
-
Pharmacokinetic
- PKPD:
-
Pharmacokinetic-pharmacodynamic
- PS:
-
Product of membrane permeability and surface area
- QTP:
-
Quetiapine
- r:
-
Rat (prefix)
- RIS:
-
Risperidone
- RO:
-
Receptor occupancy
- SPECT:
-
Single-photon emission computed tomography
References
de Greef R, Maloney A, Olsson-Gisleskog P, Schoemaker J, Panagides J. Dopamine D(2) occupancy as a biomarker for antipsychotics: quantifying the relationship with efficacy and extrapyramidal symptoms. AAPS J. 2011;13(1):121–30.
Farde L, Nordstrom AL, Wiesel FA, Pauli S, Halldin C, Sedvall G. Positron emission tomographic analysis of central D1-dopamine and D2-dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine - relation to extrapyramidal side-effects. Arch Gen Psychiatry. 1992;49(7):538–44.
Nordstrom AL, Farde L, Wiesel FA, Forslund K, Pauli S, Halldin C, et al. Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects - a double-blind pet study of schizophrenic-patients. Biol Psychiatry. 1993;33(4):227–35.
Kapur S, Remington G, Jones C, Wilson A, DaSilva J, Houle S, et al. High levels of dopamine D-2 receptor occupancy with low-dose haloperidol treatment: a PET study. Am J Psychiatr. 1996;153(7):948–50.
Horacek J, Bubenikova-Valesova V, Kopecek M, Palenicek T, Dockery C, Mohr P, et al. Mechanism of action of atypical antipsychotic drugs and the neurobiology of schizophrenia. CNS Drugs. 2006;20(5):389–409.
Grimwood S, Hartig PR. Target site occupancy: emerging generalizations from clinical and preclinical studies. Pharmacol Ther. 2009;122(3):281–301.
Johnson M, Kozielska M, Reddy VP, Vermeulen A, Li C, Grimwood S, et al. Mechanism-based pharmacokinetic-pharmacodynamic modeling of the dopamine D(2) receptor occupancy of olanzapine in rats. Pharm Res. 2011;28(10):2490–504.
Kozielska M, Johnson M, Reddy VP, Vermeulen A, Li C, Grimwood S, et al. Pharmacokinetic-pharmacodynamic modeling of the D2 and 5-HT2A receptor occupancy of risperidone and paliperidone in rats. Pharm Res. 2012;29(7):1932–48.
Danhof M, De Lange ECM, la Pasqua OE, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic (PK-PD) modeling in translational drug research. Trends Pharmacol Sci. 2008;29(4):186–91.
Garberg P, Ball M, Borg N, Cecchelli R, Fenart L, Hurst RD, et al. In vitro models for the blood–brain barrier. Toxicol in Vitro. 2005;19(3):299–334.
Uchida Y, Ohtsuki S, Katsukura Y, Ikeda C, Suzuki T, Kamiie J, et al. Quantitative targeted absolute proteomics of human blood–brain barrier transporters and receptors. J Neurochem. 2011;117(2):333–45.
Mager DE, Jusko WJ. Development of translational pharmacokinetic-pharmacodynamic models. Clin Pharmacol Ther. 2008;83(6):909–12.
Attarbaschi T, Geiss-Granadia T, Sacher J, Klein N, Mossaheb N, Wiesegger G, et al. Striatal D2 receptor occupancy in bipolar patients treated with olanzapine. Biol Psychiatry. 2005;57(8):169S.
Dresel S, Mager T, Rossmuller B, Meisenzahl E, Hahn K, Moller HJ, et al. In vivo effects of olanzapine on striatal dopamine D-2/D-3 receptor binding in schizophrenic patients: an iodine-123 iodobenzamide single-photon emission tomography study. Eur J Nucl Med. 1999;26(8):862–8.
Nordstrom AL, Farde L, Nyberg S, Karlsson P, Halldin C, Sedvall G. D-2 receptor occupancy in relation to clozapine serum concentration - a pet study in schizophrenic-patients. Psychopharmacology (Berlin). 1995;118(2):B8.
Nordstrom AL, Farde L, Halldin C. Time course of D2-dopamine receptor occupancy examined by pet after single oral doses of haloperidol. Psychopharmacology (Berlin). 1992;106(4):433–8.
Lavalaye J, Linszen DH, Booij J, Reneman L, Gersons BPR, van Royen EA. Dopamine D-2 receptor occupancy by olanzapine or risperidone in young patients with schizophrenia. Psychiatry Res-Neuroimaging. 1999;92(1):33–44.
Nordstrom AL, Nyberg S, Olsson H, Farde L, Seeman P. Positron emission tomography finding of a high striatal D2 receptor occupancy in olanzapine-treated patients. Arch Gen Psychiatry. 1998;55(3):283–4.
Nyberg S, Farde L, Halldin C. A PET study of 5-HT2 and D-2 dopamine receptor occupancy induced by olanzapine in healthy subjects. Neuropsychopharmacology. 1997;16(1):1–7.
Pilowsky LS, OConnell P, Davies N, Busatto GF, Costa DC, Murray RM, et al. In vivo effects on striatal dopamine D-2 receptor binding by the novel atypical antipsychotic drug sertindole - A I-123 IBZM single photon emission tomography (SPET) study. Psychopharmacology (Berlin). 1997;130(2):152–8.
Tauscher J, Kufferle B, Asenbaum S, Fischer P, Pezawas L, Barnas C, et al. In vivo I-123 IBZM SPECT imaging of striatal dopamine-2 receptor occupancy in schizophrenic patients treated with olanzapine in comparison to clozapine and haloperidol. Psychopharmacology (Berlin). 1999;141(2):175–81.
Xiberas X, Martinot JL, Mallet L, Artiges E, Loc’h C, Maziere B, et al. Extrastriatal and striatal D-2 dopamine receptor blockade with haloperidol or new antipsychotic drugs in patients with schizophrenia. Br J Psychiatry. 2001;179:503–8.
Hagberg G, Gefvert O, Bergstrom M, Wieselgren IM, Lindstrom L, Wiesel FA, et al. N-[C-11] methylspiperone PET, in contrast to [C-11] raclopride, fails to detect D-2 receptor occupancy by an atypical neuroleptic. 1998;82 Suppl 3:147–60.
Arakawa R, Ito H, Takano A, Takahashi H, Morimoto T, Sassa T, et al. Dose-finding study of paliperidone ER based on striatal and extrastriatal dopamine D2 receptor occupancy in patients with schizophrenia. 2008;197 Suppl 2:229–35.
Catafau AM, Penengo MM, Nucci G, Bullich S, Corripio I, Parellada E, et al. Pharmacokinetics and time-course of D(2) receptor occupancy induced by atypical antipsychotics in stabilized schizophrenic patients. J Psychopharmacol. 2008;22(8):882–94.
Vermeulen A, Piotrovsky V, Ludwig EA. Population pharmacokinetics of risperidone and 9-hydroxyrisperidone in patients with acute episodes associated with bipolar I disorder. J Pharmacokinet Pharmacodyn. 2007;34(2):183–206.
Fagerholm U. The highly permeable blood–brain barrier: an evaluation of current opinions about brain uptake capacity. Drug Discov Today. 2007;12(23–24):1076–82.
Rengachary SS, Ellenbogen RG. Principles of neurosurgery. Edinburgh: Elsevier Mosby; 2005.
Yin DL, Valles FE, Fiandaca MS, Forsayeth J, Larson P, Starr P, et al. Striatal volume differences between non-human and human primates. J Neurosci Methods. 2009;176(2):200–5.
Farde L, Hall H, Pauli S, Halldin C. Variability in D-2-dopamine receptor density and affinity - a pet study with [C-11] raclopride in man. Synapse. 1995;20(3):200–8.
Pazos A, Probst A, Palacios JM. Serotonin receptors in the human-brain. 4. Autoradiographic mapping of serotonin-2 receptors. Neuroscience. 1987;21(1):123–39.
Summerfield SG, Read K, Begley DJ, Obradovic T, Hidalgo IJ, Coggon S, et al. Central nervous system drug disposition: the relationship between in situ brain permeability and brain free fraction. J Pharmacol Exp Ther. 2007;322(1):205–13.
Summerfield SG, Lucas AJ, Porter RA, Jeffrey P, Gunn RN, Read KR, et al. Toward an improved prediction of human in vivo brain penetration. Xenobiotica. 2008;38(12):1518–35.
Liu XR, Van Natta K, Yeo H, Vilenski O, Weller PE, Worboys PD, et al. Unbound drug concentration in brain homogenate and cerebral spinal fluid at steady state as a surrogate for unbound concentration in brain interstitial fluid. Drug Metab Dispos. 2009;37(4):787–93.
Keck PE, McElroy SL. Clinical pharmacodynamics and pharmacokinetics of antimanic and mood-stabilizing medications. J Clin Psychiatry. 2002;63:3–11.
Mannens G, Meuldermans W, Snoeck E, Heykants J. Plasma-protein binding of risperidone and its distribution in blood. 1994;114 Supll 4:566–72.
Feng B, Mills JB, Davidson RE, Mireles RJ, Janiszewski JS, Troutman MD, et al. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab Dispos. 2008;36(2):268–75.
Kapur S, Seeman P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatry Neurosci. 2000;25(2):161–6.
Seeman P. Atypical antipsychotics: mechanism of action. Can J Psychiatry. 2002;47(1):27–38.
Kassahun K, Mattiuz E, Nyhart Jr E, Obermeyer B, Gillespie T, Murphy A, et al. Disposition and biotransformation of the antipsychotic agent olanzapine in humans. Drug Metab Dispos. 1997;25:81–93.
Sun HD, Pang KS. Permeability, transport, and metabolism of solutes in caco-2 cell monolayers: a theoretical study. Drug Metab Dispos. 2008;36(1):102–23.
Kwon H, Lionberger RA, Yu LX. Impact of P-glycoprotein-mediated intestinal efflux kinetics on oral bioavailability of P-glycoprotein substrates. Mol Pharm. 2004;1(6):455–65.
Schotte A, Janssen PF, Gommeren W, Luyten WH, Van GP, Lesage AS, et al. Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berlin). 1996;124(1–2):57–73.
Seeman P, Tallerico T. Antipsychotic drugs which elicit little or no Parkinsonism bind more loosely than dopamine to brain D2 receptors, yet occupy high levels of these receptors. Mol Psychiatry. 1998;3(2):123–34.
R Development Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009.
Kapur S, Zipursky RB, Remington G. Clinical and theoretical implications of 5-HT(2) and D(2) receptor occupancy of clozapine, risperidone, and olanzapine in schizophrenia. Am J Psychiatr. 1999;156(2):286–93.
Hammarlund-Udenaes M, Friden M, Syvanen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25(8):1737–50.
Avdeef A. How well can in vitro brain microcapillary endothelial cell models predict rodent in vivo blood–brain barrier permeability? Eur J Pharm Sci. 2011;43(3):109–24.
Danhof M, de Jongh J, De Lange ECM, la Pasqua O, Ploeger BA, Voskuyl RA. Mechanism-based pharmacokinetic-pharmacodynamic modeling: biophase distribution, receptor theory, and dynamical systems analysis. Annu Rev Pharmacol Toxicol. 2007;47:357–400.
Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation. Br J Pharmacol. 2010;161(6):1219–37.
Zhang G, Terry AV, Bartlett MG. Sensitive liquid chromatography/tandem mass spectrometry method for the simultaneous determination of olanzapine, risperidone, 9-hydroxyrisperidone, clozapine, haloperidol and ziprasidone in rat brain tissue. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858(1–2):276–81.
Chang WH, Lin SK, Jann MW. Interconversions between haloperidol and reduced haloperidol in schizophrenic-patients and Guinea-pigs - a steady-state study. J Clin Psychopharmacol. 1991;11(2):99–105.
Kalvass JC, Pollack GM. Kinetic considerations for the quantitative assessment of efflux activity and inhibition: implications for understanding and predicting the effects of efflux inhibition. Pharm Res. 2007;24(2):265–76.
ACKNOWLEDGMENTS AND DISCLOSURES
Martin Johnson is an employee of Astrazeneca, Cambridge, UK. Venkatesh Pilla reddy is an employee of Astrazeneca, Cambridge, UK and holder of AstraZeneca shares. This research article was prepared within the framework of project no. D2-104 of the Dutch Top Institute Pharma (Leiden, the Netherlands; www.tipharma.com). The authors have no conflicts of interest that are directly relevant to the contents of this research article.
Author information
Authors and Affiliations
Corresponding author
Appendices
APPENDIX 1
Derivation of the relationship between CLbev, CLeff, Papp, SA and ER
1. Relationship between CLbev, CLeff, PS and PSpgp
The derivation is based on the model described by Kwon et al. (42). The model describes the passive and active transport from the apical side (A) to the basal side (B) of enterocytes. A similar model was described by Kalvass et al. (53) for various situations where efflux transporters such as P-glycoprotein (Pgp) are involved, including the blood–brain barrier.
Equation (Eq 2) of Kwon et al. (42) describes the net change of the amount in the cells (M), i.e., the rate of accumulation in the cell.
where CA and CB are the extracellular concentration at the apical and basal side, respectively, Cent is the intracellular concentration, PS1, PS2, PS3 and PS4 are the products of membrane permeability and surface area for passive transport from A to intracellular, from intracellular to A, from intracellular to B and from B to intracellular, respectively, and PSpgp is the product PS for active transport from intracellular to A by the transporter Pgp.
In situations where the concentrations at both sides A and B are constant, Cent will be constant, and the net change of M will be zero. For dM/dt = 0, Eq. (Eq 5) can be rearranged and simplified to:
The net transport over the basal membrane in the direction from apical to basal side is
Replacing Cent by Eq. (A1)
The net transport over the apical membrane in the direction from apical to basal side is
Replacing Cent by Eq. (A1) yields the same result as Eq. (A3), since it was assumed that there is no accumulation in the cells.
The passive diffusion clearance CLbev is defined as the passive membrane flux from A to B divided by the concentration CA, so it follows from Eq. (A3):
The active efflux clearance CLeff is defined as the efflux by Pgp of CB, so from Eq. (A3):
Equations (A5) and (A6) define the relationship between the model parameters CLbev and CLeff, and their determinants, PS and PSpgp.
Note that the passive CLbev is dependent on PSpgp. This is due to the fact that the active efflux lowers the intracellular concentration Cent, reducing the effective concentration gradient over the basal membrane. As a result, the passive transport is lower compared to the situation in the absence of active efflux.
2. Efflux Ratio
The Efflux Ratio is defined as the ratio of the transport clearance (CLBA) from B to A if CA = 0 and the transport clearance (CLAB) from A to B if CB = 0 (37). From Eqs. (A3), (A5) and (A6) the following equations can be derived:
From Eqs. (A3) and (A7) it can be derived that the ratio CA/CB (plasma/brain ratio) at steady state is equal to the efflux ratio ER.
3. Relationship between Papp and PS
The apparent permeability Papp is determined by measuring the net flux over the cell layer in the absence of active efflux (PSpgp = 0). In this case Eq. (A3) can simplified to:

Papp is obtained by dividing by the concentration gradient, after normalizing for surface area (SA):
Or
The factor 2 reflects the passage over two membranes in series.
4. Relationship between CLbev, CLeff, Papp and ER
Combining Eqs. (A5), (A6), (A9), (A10) and (A13) yields the following equations:
Equations (A14) and (A15) can be used to calculate the model parameters CLbev and CLeff from the experimentally obtained values Papp and ER.
APPENDIX 2
PBPKPD model used to predict human D2RO is defined by the following differential equations:
Where,
subscripts plasma, bv, bev, stf, sb represent volume (V) and amount (A) of drug in plasma, brain-vascular, brain-extravascular, striatum-free and striatum-bound compartments, respectively;
CLbv, CLbev, CLst represent transport clearance of drug to brain-vascular, brain-extravascular, striatum-free compartments, respectively;
CLeff represents the active efflux transport clearance of drug from brain-extravascular compartment;
Bmax is the dopamine D2 receptor density in nM;
CB is the concentration bound to receptor as (Astb/Vstb)/ (MW/1000) in nM;
MW is the molecular weight of the drug;
D2RO is calculated as CB/Bmax*100%.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Johnson, M., Kozielska, M., Pilla Reddy, V. et al. Translational Modeling in Schizophrenia: Predicting Human Dopamine D2 Receptor Occupancy. Pharm Res 33, 1003–1017 (2016). https://doi.org/10.1007/s11095-015-1846-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1846-4