
Abstract
Lipid excipients are applied for numerous purposes such as taste masking, controlled release, improvement of swallowability and moisture protection. Several melting techniques have evolved in the last decades. Common examples are melt coating, melt granulation and melt extrusion. The required equipment ranges from ordinary glass beakers for lab scale up to large machines such as fluid bed coaters, spray dryers or extruders. This allows for upscaling to pilot or production scale. Solvent free melt processing provides a cost-effective, time-saving and eco-friendly method for the food and pharmaceutical industries. This review intends to give a critical overview of the published literature on experiences, formulations and challenges and to show possibilities for future developments in this promising field. Moreover, it should serve as a guide for selecting the best excipients and manufacturing techniques for the development of a product with specific properties using solvent free melt processing.
Similar content being viewed by others

Introduction
The importance of lipid-based solid oral formulations has increased during the last decades, due to their outstanding benefits such as providing modified release profiles or taste masking using solvent-free processing techniques. Lipid-based excipients were first used in the 1960s for embedding drugs in a wax matrix in order to sustain drug release (1,2). In the more recent years these excipients were successfully used in oral drug delivery systems to enhance the bioavailability of poorly aqueous soluble drugs (3–5). Furthermore, taste masking and the improvement of swallowability have been achieved with these excipients (6). Further reasons for the application of lipids in a formulation may be (I) shelf life extension by protecting the drug from other ingredients or from environmental influences (II) the reduction of gastric irritation (III) the improvement of general attributes like flowability, lubrication performance, compressibility or mechanical resistance (7,8). Common techniques to obtain solid lipid-based formulations are: spray congealing/drying, adsorption on solid carriers, melt granulation/melt extrusion, supercritical fluid based methods, or processing of solid lipid nanoparticles (9). The major difference between these methods is whether or not solvents are applied during the process. Several formulations described in the literature are prepared by spraying techniques involving lipid excipients emulsified in water or dissolved in organic solvents and also aqueous dispersions of solid lipid nanoparticles (9–12). However, there are several drawbacks associated with the presence of solvents. An aqueous medium requires a time-consuming solvent evaporation step and residual moisture may affect product stability due to drug hydrolysis. Organic solvents offer a considerably faster evaporation step, though these solvents are much more expensive, flammable and often toxic. This causes other issues like the prevention of hazards to employees and environment, and additional costly steps for solvent recovery and disposal (13). On the contrary, working with molten excipients provides the outstanding benefit of evading any solvent. Thereby, all the described disadvantages can be overcome. Despite the promising benefits, there are also some problems involved with the application of solvent-free melting techniques with lipid based excipients, e.g., heat sensitive drugs may undergo degradation and polymorphic and morphological changes of the lipid or crystallinity changes of the drug may alter the product performance over time.
Countless studies on the development of solid oral formulations using solvent-free melting techniques have been published in the last decades. The current work is a critical review of these studies based on the applied excipients, processing strategies and the analytical methods for the characterization of the excipients and the end product. The focus is on manufacturing processes, which are categorized into three main strategies, i.e., melt extrusion, melt coating and melt agglomeration. Additionally, this review intends to give support for selecting appropriate lipid excipients and suitable solvent-free manufacturing techniques to obtain the desired end product.
Lipid-Based Excipients
Substances containing a fatty acid in their chemical structure are referred to as lipid-based excipients. Even though fatty acids occur naturally and are part of the daily diet, a general statement about the safety and toxicity of their derivatives cannot be made. A reasonable approach is the selection of lipid-based excipients listed as “generally recognized as safe” (GRAS) or registered as inactive ingredient in the “inactive ingredient guide (IIG)” published by the United States Food and Drug Administration (FDA) or the use of food excipients, which only require a simplified authorization procedure for pharmaceutical application (14–16). However, physiological and biopharmaceutical interactions with other formulation compounds as well as additional toxicological effects of the formulation are not taken into account (14,17). Many lipophilic compounds act as substrates for the P-glycoprotein (PgP), a membrane-bound ATP-dependent efflux pump in the enterocyte that transports xenobiotics back into the intestinal lumen, and for the intestinal cytochrome P450 enzyme, a hydroxylase that metabolizes xenobiotics to more water soluble, often inactive derivatives that can be excreted through the urinary bladder (18,19). Their inhibition increases the absorption of drugs out of the intestinal lumen and reduces the metabolization into inactive drug derivatives leading to an overall higher drug exposure, which may go along with more pronounced side effects (18). An in vitro study, for instance, revealed that lipid-based additives such as Peceol and Gelucire 44/14, both approved as GRAS, inhibit the PgP-mediated efflux in human colon adenocarcinoma cells (Caco-2) by decreasing the PgP-protein expression (20). However, the prediction of the real in vivo impact is hardly possible, as it strongly depends on the applied excipient concentration, the binding-affinity, the therapeutic index of the drug, and even on the individual genetic protein polymorphism (18,21).
Nevertheless, the number of possible combinations of the molecular structure is virtually infinite, depending on the fatty acid (e.g., chain length, the grade of saturation, the presence of branches, the kind of chemical bond (e.g., ether/ester) and nature and number of the linked molecule (e.g., glycerol, additional fatty acids, propylene glycol, polyglycerol, sucrose etc.). This leads to a wide field of application possibilities of these excipients comprising solubility enhancement as well as controlled drug release. A useful indicator to estimate the drug release is the hydrophilic-lipophilic balance (HLB) a numerical rating scheme based on the water solubility and polarity of the excipient (22,23). As the contact angle and, consequently, the wetting behavior are affected by the HLB, excipients with a lower HLB (≤5) can be expected to retard the original drug dissolution pattern (24). Apart from polarity the gel-formation properties, melting point, crystallinity and porosity as well as the nature of the drug (e.g., solubility, melting point) itself and the way the drug is integrated into the formulation (e.g., core/shell, matrix), can play an integral role in in vitro drug release (24–26). Table I gives an overview of the substance classes and associated materials applied as excipients in published formulations for both pharmaceutical and food supplementary products. The most frequently, purely used excipients in the recent past are Compritol 888 ATO for controlled release, Precirol ATO 5 (depending on coating amount and drug (27,28)) for taste masking, Gelucire 50/13 for solubility enhancement of poor soluble drugs, and stearic acid for pH-dependent release (e.g., enteric coating). In particular, Precirol ATO 5 and Gelucire 50/13 are prone to exhibit polymorphic changes and have therefore been associated with storage instabilities. “Strategies for controlling polymorphism and formulation stability” lists a few strategies to circumvent this issue.
Common surfactants comprising a fatty acid in their molecular structure such as polyoxyethylene sorbitan fatty acid esters (Tween), sorbitan fatty acid esters (Span) or glyceryl esters of organic acids (E472 a-c) are excluded from the list. Waxy materials such as microcrystalline wax (hydrocarbon wax), carbowax and all other available polyethylene glycols are also excluded, as these materials do not comprise lipid-based structures. Nevertheless, these materials have been frequently processed with the within described melting techniques.
Characterization Methods, Challenges and Controlling Strategies
Lipid-based excipients often involve acylglycerols in their structure or their blend. Acylglycerols exist in up to four different crystalline structures: the pseudo-hexagonal sub α-, the hexagonal α-, the orthorhombic β′- and the triclinic β-form (in order of increasing thermodynamic stability) (174–177). These forms not only differ in their packing density and stability, but also in their physico-chemical properties such as their melting point, recrystallization rate, and solubility in water (177–179). For instance, the transformation from the thermodynamically instable and less dense α-form to the most stable and densely packed β-form should result in the reduction of wettability. Another effect associated with polymorphic transformation occurs during the transformation from the hexagonal α- to the triclinic β-form while passing the orthorhombic β′-form. The altered geometric integrity of the crystalline structure leads to the spontaneous formation of flake-like fractal structures on the surface also known as “blooming” (164). Interestingly, the impact of this alteration on drug release described in the literature points towards two different directions, both causing a change in drug release after storage and indicating formulation instability. On the one hand, the rough surface was described as super water-repellent with contact angles larger than 150°, meaning a significant reduction of wettability (180,181). Several studies have proven the formation of fractal structures on the surface of different lipid formulations with SEM images, which seems to be associated with an undesired retarded drug release after storage. It is of particular interest, that all described formulation comprised mono-, di- or triacylglycerides in a extruded mixture with the drug (159,182). This effect was even able to overrule the dissolution enhancing effect of some surfactants (e.g., glyceryl monostearate, HLB 3.8) (131). On the other hand, several formulations have been reported showing a significant increase in their dissolution rate after storage (80,102,183).
Interestingly, these formulations all included a compound with a high HLB and a lower melting point (e.g., Caprol PGE 860), which might form an amorphous partition in the surrounding of the crystallized lipid structures. The storage above the melting point increases the mobility of this amorphous phase and induces phase separation and may result in an increased wettability. This effect can be promoted additionally by changes in the structure of the crystalline phase due to polymorphic transition during storage (“thermally induced phase separation-recrystallization”). These phenomena are highly dependent on the characteristics of the raw material (e.g., melting point, miscibility, crystal structure) as well as on the temperature and mobility of the molecules. In particular Gelucire 50/13, which is a mixture of mono-, di- and triacylglycerides (C12-C18) and polyethylene glycol esters, is already known to show an increased release rate due to thermally induced phase separation and polymorphic transformation (184). The last example, Precirol ATO5, will emphasize, how complex and severe the impact of the polymorphic behavior on formulation stability can be. Hamdani et al. reported a complete loss of the prolonged release properties after storage at accelerated conditions of melt-granulated pellets comprising Phenylephrine, Lactose 450, Precirol ATO 5 and different amounts of unmelted Compritol 888 ATO. However, the storage at 25°C/60% r.h. decreased the dissolution rate significantly (138). Reitz et al. reported a similar impact on drug dissolution rate by using Precirol ATO 5 for manufacturing theophylline extrudates. Although the dissolution rate decreased in the first week, after 9 months it was significantly higher than directly after manufacturing (142). The mechanism behind these observations is not explained, but it is nonetheless possible that the formation of more stable polymorphs and fractal structures reduces the dissolution rate after storing at room temperature. The increase of dissolution rate observed after storage at higher temperatures lacks an explanation and has to be associated with the heterogeneous composition and sensibility to thermal treatment (141). Nevertheless, the formulation design (e.g., melting technique, excipient composition, drug properties and interaction with excipients etc.) may also affect the drug release during storage.
As the dissolution profile is influenced by various effects and complex interactions, it is not surprising that there also exist stable formulations with excipients that caused storage instabilities in other studies (139,185). Hence, it is crucial to understand the physicochemical behavior of the raw materials and the interaction and impact on the properties of the processed formulation. Therefore, the following subchapters will focus on the characterization methods for the excipients and the product after thermal processing. Subchapter 3.3 intends to show how storage instabilities may occur and how they can already be prevented in the preformulation stage.
Characterization of the Excipients
Changes in polymorphism and morphological structure as well as phase separation phenomena are probably the most common effects inducing formulation instabilities during storage. Therefore it is crucial to develop a deep understanding of the physicochemical properties of the selected excipients before and after melting, under different conditions (e.g., time, temperature), and in mixtures with each other and with and without the drug. This step saves time and costs in the process development phase. Table II summarizes methods used to gain insight into physico-chemical properties.
Characterization of the Product After Melt Processing
Apart from the well-established powder and surface-morphology characterization techniques (72,109,110,126,187,188), analytical in vitro tools are also available for the estimation of the subsequent in vivo dissolution behavior of the formulation in the body. Common in vitro methods for this purpose are dissolution tests with biorelevant media and lipolysis studies with pancreatin (182,189–193). Biorelevant media contains physiologically relevant surfactants including a mixture of bile salts (e.g., sodium taurocholate), phospholipids, pancreatic lipase to simulate lipolysis, and buffer and salts are added to adjust the pH and the osmolarity of the medium to imitate physiological conditions (182). Witzleb et al. showed that the release from lipid-based matrices in biorelevant media highly depends on the structure of the lipid. Cetyl palmitate and glyceryl monostearate, for instance, exhibited a significantly faster release in biorelevant media than in HCl due to a different solubilization with surfactants and enzymatic degradation in case of glyceryl monostearate. Other lipid matrices such as glyceryl tripalmitate and glyceryl dibehenate indicated only a minor effect of the changed medium on drug release (182). In addition, lipolysis measurements revealed that lipid digestion of the pancreatic lipase does not only depend on the chain length, but also on the grade of esterification, on the solid state, and the ability for solubilization (182,194,195). Hence, it cannot be excluded that changes in crystallinity and solubility due to polymorphic transition may have a significant impact on in vitro and in vivo drug release profiles (182). Along with an optimal dissolution rate, taste masking is often an important objective in formulation development (33,43,196,197). This is usually evaluated in sensory studies with trained volunteers (33,198–200). Some studies also involve cats, which are sensitive to bitterness and scorn bitter food with insufficient taste masking (197). A simple in vitro method is the use of a disintegration tester (1 min stirring, pH 7,4) for the evaluation of the short time release amount in a pH close to that in the mouth (196), but also the results of a test of the first minutes of dissolution have been utilized (201). Another method reported is the mini column method, which represents a release test that attempts to simulate the anatomy and physiology of the oral cavity (201,202). A more sophisticated method is the measurement of dissolved samples with an electronic tongue, which consists of different potentiometric sensors and a pattern recognition system (203,204) (Table III).
Strategies for Controlling Polymorphism and Formulation Stability
Several approaches to control lipid polymorphism are conceivable, whereby only some have been applied in the existing literature:
-
Tempering during processing:
Process temperatures are kept between the melting point of the instable α- and stable β-form, in the area where the direct recrystallization of the stable β-form predominates over the β′-form (205). This prevents the formation of fractal structures (159) and the alteration of the dissolution profile due to morphological changes during storage. The recrystallization of the thermodynamically stable β-form is significantly slower than of the α-form. This may result in processing difficulties such as agglomeration in a fluid bed melt coating process or incomplete resolidification after spray chilling in a spray dryer. Melt extrusion is an appropriate technique to generate storage stable extrudates with this technique (142).
-
Tempering after processing (“maturing”):
In the first step recrystallization of the instable α-form is enabled by ensuring optimum process conditions. Maturing is performed after the process at elevated temperatures below but close to the melting point of the instable α-form. The maturing step is time consuming and may require additional equipment (e.g., a drying chamber). Alternatively, a fluid bed coater can be used for that step, as this prevents the formation of agglomerates. The α → β transformation causes the formation of fractal structures and the alteration of the dissolution profile before and after maturing (64,66,146). A complete transformation is essential for achieving a storage stable formulation.
-
Addition of crystallization seeds (“template effect”)
The stable β-form or material with a comparable saturation grade and a chain length difference of n ≤ 4 can be used as seed material (177). The seed material is added to the molten lipid in the solid state. The presence of seeds acting as templates should accelerate the direct recrystallization of the molten material into the stable β-form (67,177). In case the stable β-form recrystallizes directly from the melt during the process, fractal structures should be avoided (159,164). When seed material is applied in form of a spraying suspension, the amount is limited to prevent nozzle clogging. The efficiency of this measure in accelerating the recrystallization and its effect on the purity of the stable phase haven’t been analyzed in detail yet.
-
Avoidance of melting:
The polymorphic transformation of most lipid excipients is monotropic. That means that if melting of the raw material, which is usually provided by the supplier in the stable β-form, can be avoided, the storage instabilities due to polymorphic changes can be eliminated. The challenge herein lies in the processability. Solid lipid excipients with a critical polymorphic behavior should either have a suitable viscosity or have to be combined with an excipient that has an appropriate viscosity and/or shows no polymorphism or transforms into the stable form during or immediately after resolidification (161,207–209). In the case of aiming at a controlled release formulation, the formation of a physical mixture between drug and lipid excipient might be the wrong path as the dissolution rate of the physical mixtures may be higher than that of the solidified melt-dispersion (140,210).
-
Addition of polymorphic modifiers:
Several emulsifiers are known to have a significant impact on the crystallization (e.g., nucleation rate, crystal growth and morphology) and polymorphic transition of lipid excipients (211,212). Although numerous publications exist on this topic, the majority of these studies discuss applying commercially available additives, which often consist of a chemical mixture with high heterogeneity and differ in their composition between different suppliers and even batches (211). Additional effects such as their concentration, kind of lipid, degree of undercooling as well as the use of agitation may have a significant influence on the crystallization and polymorphic process (211). Most of the application examples derive from and are addressed to the food industry and only a few studies are specific to the pharmaceutical industry (131,162,213,214). There are several examples how modifiers can impact the polymorphism: Sucrose esters obstruct the α → β and β′ → β transition in tristearin and hydrogenate sunflower oil due to their high rigidity and some representatives (e.g., P-170, S-170) also affect the crystal size of the high-melting fraction of milk fat (215–217). Garti et al. claimed that solid emulsifiers such as Span 40 (sorbitan monopalmitate), Span 60 (sorbitan monostearate), Span 65 (sorbitan tristearate) as well as glyceryl monostearate were able to stabilize the α-form and prevented the transformation into the stable β-form in tristearin (218,219). However, ageing experiments at room temperature revealed that only a few molecules (e.g., triglycerol-l-stearate, sorbitan monostearate) with a suitable dimension of the hydrophilic moiety (“button syndrome”) were able to preserve a certain amount of the α-form for a longer storage time (218). As the effect on polymorphism depends on the chemical and structural nature of the lipid and emulsifier, in principle it is conceivable that the stabilization of the α-form during storage may be feasible to some extent. Nevertheless, this approach is associated with a high risk of transformation into the stable β-form in particular at higher storage temperatures. A more promising approach is the addition of emulsifiers to accelerate the transformation into the stable β-form. In particular, liquid or semi-solid emulsifiers such as different polysorbates (Tween 60, 65, 80) and sorbitan monolaurate (Span 20) were proven to promote the α → β transformation due to an increase in molecular mobility (218,220). In the pharmaceutical industry this approach was used to coat N-acetylcysteine in a fluid bed coater with a coating consisting of tripalmitin and polysorbate 65 (162). As an advantage, the process temperatures can be kept to a minimum, which is preferable for drugs sensitive to heat, and the transformation and morphological changes will be complete before storage. However, it must be borne in mind that phenomena apart from polymorphism can lead to storage instabilities in particular if a liquid or semi-solid emulsifier is used in a greater amount (e.g., phase separation). Furthermore, the application of a higher amount of low-melting excipients can pose a problem if melt coating (e.g., fluid bed coating) or fast recrystallization in general is required. Thus, it is wise to take the time and effort for pre-formulation studies exploring the polymorphic and morphological behavior (e.g., crystal size, blooming, phase separation etc.) at different conditions (e.g., temperature, cooling and recrystallization rates, concentration etc.).
-
Selection of excipients without/with stable polymorphism
Waxes such as carnauba wax, bees wax and stearyl stearate are stated to be non-polymorphic materials (40,46,221), which avoid storage instabilities associated with polymorphic changes. In particular the high melting point and brittleness of carnauba wax can induce processing problems such as nozzle clogging. Carnauba wax and bees wax have been most frequently used for controlled release formulations, but also immediate release is feasible with the addition of dissolution enhancers. Polyglycerides are said to be non-polymorphic, but stable in the α-crystalline form (222,223). The amount of glycerol- and esterified fatty acid molecules contributes to the HLB and can be adjusted in a wide polarity range and oligoglycerols with a degree of polymerization of up to 10 are approved by the FDA (224). Polyglycerol bears more functional groups for modification than PEG, and therefore shows a higher adaptability to different requirements such as the melting point or viscosity. However, these materials have been overlooked by the pharmaceutical industry for a long time (224).
-
Selection of polymorphic excipients:
Lipid excipients consisting of a complex mixture of glycerol molecules with different degrees of esterification (e.g., Precirol ATO 5, Compritol 888 ATO, Gelucire 50/13 etc.) and/or of esters of different fatty acids (e.g., Precirol ATO 5) should be treated with particular caution (70,225). The high heterogeneity leads to a complex polymorphic behavior difficult to predict during melt processing and storage (137). The polymorphic behavior of Compritol 888 ATO, which mainly consists of glyceryl dibehenate, has been studied extensively with DSC, time-resolved synchrotron x-ray diffraction and infrared spectroscopy at different conditions (e.g., cooling rate, addition of pure acyl glycerides etc.) (135,226). These studies revealed that the ratio of mono-, di- and tribehenin and especially the monobehenin amount is important for the formation of a pseudohexagonal sub-α and hexagonal α-phase (226). In particular the less stable and compact sub-α-form shows a higher drug incorporation, which is desired in the preparation of solid lipid microparticles (226,227). Therefore a variation in the batch composition of the supplier as well as the mixture with excipients containing monoacylglycerides themselves can lead to changes in the equilibrium and different polymorphic behavior (226). Nevertheless, Compritol 888 ATO has been widely used in controlled release formulations and seems to be able to provide a stable release profile during storage (43,228). In contrast, the literature seems to lack polymorphic studies for Precirol ATO 5 with a similar degree of detail. Hamdani et al. mainly used DSC and powder X-ray diffraction to investigate the sensitivity of the polymorphism on thermal treatment of Precirol ATO 5 (137). The complex polymorphic behavior due to the high inhomogeneity of the mixture led to several studies that showed an altered dissolution profile (138,142). However, the higher complexity of the aforementioned materials also brings advantages such as the broader melting and recrystallization range, which leads to a better spreading and higher robustness against process changes compared to pure materials with a sharp recrystallization profile such as triacylglycerides (e.g., Dynasan 114) (142).
-
Specification of storage temperature:
Transformation kinetics and phase separation phenomena are both temperature dependent (26,82). The selection and specification of adequate storage conditions might be the last way to prevent or at least slow them down to a minimum. However, this approach is less favored by the pharmaceutical industry as patient compliance is an issue.
Selection of Lipid-Excipients and Melt Processing Techniques
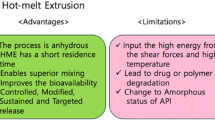
Several techniques have been described in the literature on solvent-free preparation of lipid-based solid, oral formulations. Nevertheless, the first step in formulation development should always be a careful selection of the lipid excipients (and other excipients if necessary). A useful, rationale based approach is provided by the FDA in the ICH guideline Q8 within the framework of the concept of quality by design (QbD) (229). According to this guideline a quality target product profile (QTPP) should be defined for the final or intermediate product. This QTPP should include all desired characteristics such as the dosage form, drug load, particle size, bioavailability, dissolution behavior, taste and stability (230). Based on the QTPP the next step will be the definition of the critical quality attributes (CQA) for the excipients and the drug and their appropriate specification limits, which must be met to ensure the desired product quality and patient safety (229). Typical CQAs are, for instance, the dissolution rate, the particle size distribution, the maximum permissible impurity profile (231) as well as the stability of the formulation (232). Rosiaux et al. (26) gave an overview of formulation parameters (e.g., HLB, excipient characteristics, further additives) that can be used to adjust the drug release from lipid matrices. Pore formers or surfactants with higher polarity accelerate the drug release, whereas increasing the concentration and melting point of the hydrophobic lipid excipient can be used to slow down the drug release (26). Taste masking with an immediate release profile can be achieved by applying lipids with a comparably low melting point such as Dynasan 114, Dynasan 116, Precirol ATO 5 or Witocan 42/44 (16,42,162,233–235). However, excipients with a melting point near room temperature may result in an inferior product quality due to poor flowability, a tendency to form great agglomerates and the risk of morphology changes during storage (236). Hence, comprising a liquid or a very low melting (<40°C) excipient in the formulation has to be well considered and the amount kept as low as possible. The ideal compromise between sufficient taste masking and immediate release may be found by applying a statistical design (DOE) to achieve an optimal adjustment of the critical process input parameters (CPP). Further consideration should be given to physical or chemical interactions between the chosen lipid excipients and the drug with special regard to temperature induced degradation and changes in crystallinity (solid solution, polymorphic changes). The selection of equipment and processing technique depends on the desired dosage form and particle size distribution as well as on already existing equipment and characteristics of the active ingredient and chosen excipients. The most common equipment in the industry used for melt processing are the extruder, the high shear mixer, the fluid bed, the pan coater and the spray dryer, which all allow for upscaling. Depending on the desired solid dosage form, a great number of different downstream processes are addressed in the literature, such as spheronization (49,69,109,209), molding (74,95,133,183,237,238), capsule filling (87,88,91,98,100,102,139,239–246), injection moulding (247,248), freeze pelletization (121,249), pastilation (250), milling (122,196) or tableting (92,156,251). Nevertheless, it should be obvious, that manufacturing costs will rise with the addition of further process steps and, therefore, should be considered thoroughly in advance. Table IV summarizes the common advantages and disadvantages associated with the melt processing technique in the respective equipment. A comparison with the required QTPP may provide the first support for the selection of the appropriate technique and equipment.
The following chapter will focus on the developed formulations and associated characteristics of the final product, on the basis of the categories melt extrusion, melt coating and melt agglomeration. At this point it is worthy of note that the terms for the melting techniques are not used consistently in the literature, especially when the topics melt coating and melt granulation are concerned (51). The fundamental differences between these techniques lie in the distribution of the API and the size of its contact surface with the lipid excipient. Melt extrusion provides a homogeneous solid dispersion after processing, while the distribution is less homogenous and the contact surface of the API and the lipid is smaller when melt agglomeration by high shear mixing and in particular melt coating are used. Nevertheless, the delimitation between these terms is complicated. For instance, several downstream processes of melt extrusion allow for the formation of pellets and granules, and with melt coating techniques a drug / lipid dispersion may be sprayed on nonpareils creating a solid dispersion layering.
Melt Extrusion
The classic equipment for the melt extrusion process is the extruder, which consists of a single- or twin-screw system. In both systems the screw is positioned in the center of a heatable stationary barrel. Three zones within the extruder are named after their individual function.
-
1.
Feed zone: maximum pitch between barrel and screw flight
The feed material enters this zone through a gravimetric or volumetric hopper, is mixed under low pressure and transported by the screw rotation to the compression zone.
-
2.
Compression zone: continuous reduction of the pitch along the screw
The steady pressure increase leads to compression, particle size reduction and more effective shearing and mixing. The heated barrel wall and the frictional heat are used to melt or at least soften the material. Additional equipment such as conveying and kneading elements may be used for the mixing, homogenization, venting and kneading process.
-
3.
Metering zone: constant minimum pitch
The continuous high pressure supports a homogenous and steady material flow to and through the die.
After melt extrusion the product is cooled and shaped downstream in further processing units, such as hot strand cutters, calendaring, chill rolls, or spheronizers/marumerizers (128,280,281,290 294).
Melt Extrusion is widely used for two main purposes, namely enhancing the bioavailability of poorly soluble drugs and the development of formulations with modified release profiles (294–296). Jannin et al. (9) and Keen et al. (247) reviewed a number of formulations showing an improved drug release. The preferred lipid excipient was Gelucire 44/14, which was extruded in a blend with 17 β-estradiol and PVP, PVA, or PEG 6000 (92). Mehuys et al. extruded Propranolol with HPMC and Gelucire 44/14, which was molded as core material in an ethyl cellulose pipe. The ethyl cellulose pipe offers protection against hydrodynamic and mechanical stress and provides a sustained, zero-order, erosion-controlled drug release and a better bioavailability in dogs compared to a commercial product (237,297,298). The application of extruded wax matrices for the retardation of drug release started in the early 1990s (299,300). Sato et al. and Miyagawa et al. conducted a study to analyze the influence of dissolution rate-controlling agents on extruded carnauba wax matrices with diclofenac as model drug. Their investigations highlighted the importance of a proper pre-selection of the dissolution modifiers and their physiochemical properties (301,302). Liu et al. prepared tablets from sustained-release extrudates of phenylpropanolamine, wax and different types of fillers and showed the effect on the dissolution rate caused by the type of filler in the matrix (303). This matches the investigations of De Brabander et al., who described a different impact on dissolution with different starch derivatives in extrudates compressed to mini tablets (304). Quintavalle et al. developed a sustained co-extrudate with an inner hydrophilic core (PEG 6000) and an outer lipophilic coat both containing theophylline as drug. Its release was tailored through a suitable selection of parameters. In addition a very small in vivo bioavailability study of four healthy volunteers confirmed the desired sustained release (305). Roblegg et al. introduced vegetable calcium stearate for preparing sustained release pellets with a 20% paracetamol drug load. Although the addition of dissolution enhancers like glyceryl monostearate and tributyl citrate was able to reduce the process temperatures, the temperature of 70°C is still inappropriate for thermolabile substances (128). Vithani et al. successfully applied Compritol® 888 ATO as an excipient for the extrusion of the model drug diclofenac sodium and subsequent direct compression of sustained release tablets (306). Several investigations from the working groups of Breitkreutz and Kleinebudde et al. delivered insight into the applicability of different lipid excipients and the polymorphic behavior after melt extrusion and storage (43,131,142,159,161,163). Their storage stability studies revealed a superior stability of homogenous lipids (e.g., Dynasan 114) and emphasized the requirement for understanding and avoiding polymorphic transformation during storage (43,142). With Witocan 42/44 melt extrusion at room temperatures (“solvent-free cold extrusion”) and a stable release profile over storage was feasible (42,43). Another approach for reduction of the process temperature and extending the release was adopted by Schulze and Winter for a protein co-lyophilisate of INF α with hydroxypropyl-β-cyclodextrin in a lipid implant. The selective melting of a low melting lipid excipient in a blend with a high melting lipid provided an extrudable mass at moderate temperatures, an unchanged protein structure and avoided polymorphic changes of the high melting lipid due to recrystallization (207). In a second study with lysozyme Sax and Winter chose different low melting lipids in the combination with Dynasan 118 and different portions of pore forming agent to analyze the influence on the release kinetics of protein molecules from the lipid implant (45). The partial melting of the low melting fraction at incubation temperatures of 37°C built a protein reservoir and an additional non-aqueous diffusion pathway besides the conventional way through the buffer filled interconnected pore-network created by the dissolution of hydrophilic excipients (45,165). Recently, Oliveira et al. evaluated the extrudability of formulations, comprising Gelucire 33/01 and two further lipid excipients such as Dynasan 114, Precirol ATO 5, Witocan 42/44 or Compritol 888 ATO at room temperature and revealed a complex influence on porosity, thermal behavior, mechanical properties and dissolution rate depending on the chosen composition (307). It can be concluded that if lipid excipients are used, not only the dissolution rate but also mechanical properties and porosity should be monitored during storage. A list of formulations developed via extrusion and their characteristics are summarized in Table V.
All in all, lipid-based excipients usually show a lower melt viscosity and melting point compared to polymeric excipients typically used in hot melt extrusion (e.g., hydroxypropyl methylcellulose, cellulose acetate, polymethacrylate) (294). Hence, with lipid excipients the addition of plasticizers (e.g., PEGs, triacetin, citric acid, GMS etc.) to decrease the glass transition point and melt viscosity is dispensable. This leads to a reduction of formulation complexity and allows for lower process temperatures (294,308,309). Thus, lipid excipients are able to extend the portfolio of suitable APIs for hot melt extrusion by also including heat sensitive candidates such as proteins (207). Another important issue in the recent years was the adjustment of the dissolution rate. Most of the published studies focus on sustained release formulations (128,142,303,306,310) but only a few on the application for taste masking by maintaining the immediate release profile (42,43,233,235) and enhancement of the solubility of poorly water-soluble drugs (92). The enhancement of the release profile was usually performed by adding rather high amounts of additives such as polyethylene glycol (161), or certain amounts of polymers such as PVP (92). However, the literature seems to lack in successful solvent-free melt extrusion approaches with lipid-based excipients alone, although hot melt extrusion with polymers is one of the most popular methods to enhance solubility (281). The reasons for the reluctant development of these kinds of formulations are multifactorial. The choice of lipid-based excipients for oral application suitable for this purpose is rather limited. The idea of including polyethylene groups is perhaps sufficient to increase the HLB and solubility, but leads to low melting temperatures (Gelucire 44/14—HLB 14, melting point 44°C) impeding melt processing (e.g., in spheronization step) (92) and deteriorating formulation stability. Additionally, changes in polymorphism may occur and alter dissolution rates during storage (e.g., Gelucire 50/13—HLB 13, melting point 50°C) (311). Also other excipients such as sucrose esters, which would be stable up to 140°C (312) and are available with a promising high HLB of 16, showed polymorphic transformation during storage after melting (173). Polyglycerol esters crystallize in hexagonal subcells, exhibit no polymorphic transformation, are thermally stable below 100°C (312) and available in a HLB range from 6 to 11 (313). However, only one study published in 1993 could be found where these excipients were successfully applied in a spray chilling process (314).
Melt Coating
The first publication mentioning the term “melt coating” or more precisely “hot melt coating” was an US patent published in the year 1942 (315). A solvent-free coating technology was described to coat sheet material (e.g., paper, foil, metal) with molten thermoplastic resins, which can be used for the manufacturing of water- and grease-proof primary packaging of food (315,316). In the following decades the coating with molten lipid-based excipients became a standard procedure in the food industry (317,318). The literature discusses a wide range of food products coated by recrystallization of lipid-based molten excipients. For example: dip-coating of frozen meat (“larding”) (317,319,320), spray- or dip-coating as well as enrobing of ice cream with molten chocolate (321), spray chilling/congealing or fluidized bed coating of supplements (e.g., vitamin C) (322), food additives (e.g., aspartame) (323), flavors (324), or even heat sensitive probiotic bacteria (47,259), to name just a few. The advantages of this coating method have not gone unnoticed by the pharmaceutical industry. In the 1960s spray chilling was used for the first time by Merck & Co Inc. to achieve taste masking for iron particles (325) as well as for water-soluble vitamins (326). In 1968 Richardson Merrell Inc. applied sprays of molten resin mixtures on a tumbling bed of tablets in a pan coater (327,328), which marked the birth of the so called pan spray coating procedure. This technique was also transferred to a fluid bed coater, which was equipped with an additional heating system for the atomization air by Glatt Air Techniques Inc. in the early 1990s (62, 329). This was the start of further research and development in this field, which has been reviewed in several articles (13,253,263,264,330,331). The fluidized bed coater, the pan coater and also the spouted bed are common machines used for hot melt coating (264,332–334). In a coating pan the molten material is either sprayed by a nozzle (pan spray (63,327,328)) or poured onto the rolling product (pan pour coating (113,167)). The same can be performed in a fluid bed chamber by fluidizing and melting the lipid excipient together with the product (in-situ hot melt coating/ solid dispersion fluid bed hot melt coating (123,147,335)) or by spraying (hot melt spray coating (62,221)). Classically, the drug is preheated and fluidized in the fluid bed coater, but heat stable drugs can also be homogenously dispersed in the molten coating material and sprayed on fluidized nonpareils seeds (drug lipid dispersion hot melt coating (64–66)). This makes a time-consuming process step to obtain a core material with appropriate particle characteristics (e.g., size distribution, flowability, friability etc.) superfluous. Nevertheless, this method is only suitable for a small drug load as the solid amount in the spray dispersion can clog the nozzle during spraying. Drug-lipid interactions have to be evaluated in advance, as the drug may be dissolvable in the lipid matrix and can be present in the amorphous state after resolidification (65) posing a potential risk of formulation instability due to recrystallization during storage. Further, the particle size of the drug can have a significant impact on drug release (65) and therefore has to be observed during formulation development. However, pouring techniques and in-situ hot melt coating are used more often in the context of research than in large-scale manufacturing. The use of pouring techniques in pan coating requires a high degree of attention and care, as poor execution may cause twinning as well as inferior coating homogeneity and efficiency (52). In-situ hot melt coating can be used without any additional hot-melt equipment (e.g., nozzle, heating system etc.), which may be advantageous for faster pre-studies. Nevertheless, the applicability in production scale is questionable, as core materials smaller than 420 μm are prone to agglomerate, porosity and particle size distribution affect batch-to-batch reproducibility and the lipid amount successfully applicable without agglomeration is rather low (<5%) and fails to reach the target specification (335). Hot melt spray coating requires more experience for the parameter selection, additional heating equipment, excipients with appropriate physicochemical characteristics (e.g., melting point, viscosity etc.) for the spraying process, and more time for the spraying step. Nevertheless, it is preferred over in-situ melting as it offers the option of fine adjustment of the product quality due to a higher number of process parameters, a lower risk of agglomeration, minor thermal stress of the API due to significant shorter exposure times to the molten coating, and easier up-scaling. Table VI provides a selection of formulations produced by spray coating and their intended purpose found in the literature.
Hot melt coating by direct blending is another method more recently mentioned in the literature (338,339). Drug granules or nonpareils are stirred in the molten coating or coating drug dispersion until homogeneity is reached and then congealed by cooling down to room temperature under continuous, vigorous stirring (37,339). A zero-order release was obtained by double coating of extruded acetaminophen pellets with carnauba wax and HPMC in the inner and Syncrowax HR-C (glyceryl tribehenate) in the outer coating. HPMC and EC were described as useful swelling and eroding agents, which are able to decrease initial burst release by hindering drug diffusion (338). Further, sugar spheres with a coating comprising a waxy excipient with a low melting point and higher polarity (e.g., cetostearyl alcohol, Polawax, and 1-Monostearin) and a low drug load of acetaminophen exhibited an immediate release profile. At the first glance, the processing seems very convenient and easily controllable for particles with an appropriate size (250–595 μm) without the requirement of any sophisticated equipment in laboratory scale (37), but if and how this method is suitable for up-scaling has yet to be evaluated. It will also be of further interest, if taste masking can be attained by this approach, how the dissolution profile can be tailored by different pellet and coating compositions (e.g., other drugs, higher drug load, multi-layers) and most importantly if the obtained formulations are stable during storage.
As already stated in the subchapter on hot melt extrusion, controlled release was the main application purpose for hot melt coating with lipid-based excipients. Rosiaux et al. gave a comprehensive insight into the literature on drug release mechanisms of sustained release lipid matrices (26). Immediate release was achieved with a homogenous mixture of a lipid excipient with a rather low HLB (HLB ≤ 5), a higher melting point (≥60°C), a small recrystallization range (≤35°C) and a hydrophilic polymer or emulsifier with sufficient miscibility in the melt (e.g., hydroxyethyl cellulose, hydroxypropyl cellulose, carbomer, PEG-derivatives, E-400–E-499 etc.) (40,336). The emulsifiers and disintegrants were mainly selected on the basis of their HLB and hydrophilic behavior, but only one study is known in which the emulsifier was used as a polymorphic modifier to gain the stable β-phase during and directly after the process (162). Although studies on storage stability are the most crucial point while working with lipid based formulations, in particular if complex coating compositions are used, only very few studies have taken this point into account (40,46,162). The same holds true for the evaluation of taste masking, which often is a further target of this kind of formulation. Sufficient taste masking is often assumed on the basis of the release profile in the first minute of dissolution, but only rather seldom volunteers or more sophisticated analytical tools are used for the evaluation (40).
Melt Agglomeration
Melt agglomeration includes melt granulation and melt pelletization, which provides highly spherical agglomerates with a narrow size distribution and a particle size from 0.5 to 2 mm. However, both terms cannot be clearly distinguished and are not used consistently in the published literature (340,341). Apart from using a melt extruder equipped with the necessary accessories, the formation of granules and pellets can be performed in a fluid bed (84,124,342,343) or high shear mixer (55,75,81,99,130,148,169,344). Schaefer et al. gave a deep insight into the important process parameters especially for the high shear mixer and described the following phases and mechanisms during agglomeration (125,345–350).
-
1.
Nucleation phase (345,346,351,352):
In the first step the solid lipid binder is either filled into the equipment together with the starting material consisting of excipients (e.g., filler, disintegrants etc.) and/or drug or molten externally with or without the drug. In case the binder is molten during mixing with the remaining compounds the method is described as “melt-in” (high shear mixer) or also “in-situ” (fluid bed) agglomeration. If the binder is added in the molten form it can either be sprayed with a nozzle onto the preheated material in the “spray on”-method or steadily pumped onto the preheated material in the “pour-on” or “pump-on” method. Two different nucleation mechanisms after wetting of the material have been postulated. One is the distribution mechanism where the molten binder spreads over the surface of the starting material and primary nuclei are formed by coalescence. The other is the immersion mechanism, which is predominant if the molten binder droplets are larger than the starting material particles and the solid particles immerse into the molten droplet surface to form the nuclei. The droplet size can be affected by the shear rate (melt-in, pour-on), the initial particle size of the lipid binder (melt-in), the spray rate and pressure (spray-on), as well as by the binder viscosity at the applied temperature. The nucleation takes place as long as nuclei interact with initial particles. This leads to a depletion of fines and finally to an increased wetting and starting of growth by coalescence between nuclei.
-
2.
Growth phase (345,346,351,352):
The growth phase presents an equilibrium between consolidation and growth until a critical size is reached and attrition and breakage into smaller particles takes place. The critical size depends on several parameters such as the physicochemical properties and amount of the binder (e.g., particle size, viscosity, deformability, temperature etc.), characteristics of the starting material (particle size, shape, density etc.) and kinetic energy applied (impeller speed and frictional heat, air temperature) (346,351,352). Parameters such as the particle size and shape, the binder viscosity and a certain solid/lipid ratio also have an effect on the granule strength and whether agglomerates are formed and densified by coalescence or if breakage to fragments and layering on the existing agglomerates is present. In case the binder viscosity and lipid/solid ratio are well-chosen, densification and a steady growth will take place and depending on the binder amount and the applied shear force, particle size distribution narrows, sphericity increases and porosity decreases (melt pelletization).
-
3.
The cooling phase can be performed by cooling the particles in the equipment, which is much faster in the fluid bed than in the high shear mixer due to the better heat transfer, or by rapid cooling by pouring the material directly into liquid nitrogen (“flash-cooling”) or by simply spreading the material out in thin layers on trays (high shear mixer). In case of polymorphic material or material that can vary in the degree of its crystallinity (e.g., PEG 3000), the cooling rate can play an essential role in controlling formulation stability and drug release (81). Table VII lists a selection of formulations prepared by melt agglomeration in a high shear mixer and fluid bed.
Table VII Selection of Formulations Produced by Melt Agglomeration Techniques
Several designs have been applied to understand the granulation mechanisms and the effect of different process parameters on product quality (71,99,118,198,274,357). Comparison studies using in-line particle size measurement tools (focused beam reflectance measurement, spatial filter velocimetry) for the endpoint determination between the binder application methods (melt-in/in-situ, spray-on) revealed that the results were comparable in respect of particle size distribution and flowability. The dissolution profiles of the granules processed in a fluid bed and in a high shear mixer were comparable, provided the drug was applied in the same close contact to the lipid excipients (126,277). Further studies revealed that both fluid bed methods, spray-on as well as in-situ, were able to obtain smooth and spherical granules with a comparably fast dissolution rate for granules with the same size, while the particle size distribution seems to be positively affected by smaller binder particles or droplets in both methods (354). Studies revealed that melt agglomeration in a fluid bed can be successfully applied to improve the lubricant performance, flowability, and compressibility of the granules in a subsequent tableting step (358,359). This is caused by a homogenous repartition and due to the significantly lower shear force in a fluid bed than in a high shear mixer that leads to a reduction of particle densification and to an increased elastic deformation and better compressible granules (124). Moreover, the fluid bed offers a more efficient cooling process without additional densification, which is an advantage for up-scaling and enables the addition of higher binder contents (343). Further, Kukec et al. were able to successfully scale an in-situ laboratory method up to pilot scale by the aid of a three-factor, five-level circumscribed central composite design, whereby the resulting dissolution rate was affected mainly by the binder content (261). Hence, in-situ melt fluid bed granulation is a viable and fast method to obtain granules from moisture sensitive drugs.
Other Methods
Spray Congealing (Synonyms: Spray Chilling, Spray Cooling)
Although spray congealing is often listed as a melt agglomeration technique (340,352,360), it can be seen as a mix of all three aforementioned methods, as the products are perfectly spherical microspheres or microcapsules that can contain the drug embedded as a solid dispersion in the lipid matrix. The nozzle type and fluid delivery determines whether a matrix (“microsphere”) or a more core/shell-like structure (“microcapsules”) of the drug embedment is generated. Prilling is a special form of spray-congealing that delivers a product with an increased particle size of 500 to 2000 μm (“prills”) (120,157,361). For spray congealing a stable and homogenous mixed melt dispersion of the drug (and further excipients if necessary) is required. The chosen technique (e.g., ultrasound homogenizer, high shear mixer) for the preparation of the dispersion may have a significant impact on the rheological behavior, particle size and crystallinity and should be therefore controlled during product development and processing (76). This mixture is subsequently processed in a choice of atomization units, which are distinguished by their atomization mechanism, liquid channeling, throughput and product properties (e.g., particle size and distribution, structure etc.) (285,286). Examples mentioned in the literature are the spinning disk (127,132,362,363), vibrating nozzle (157,361), pneumatic nozzle (101,120,364,365), ultrasonic devices (72, 77,78,366–368), or dual-fluid nozzle (73,76). After atomization the particles are solidified by falling in a large prilling tower, a chamber of a spray dryer flushed with cold air, liquid nitrogen or cooled by a carbon dioxide ice bath (285,364). This step is critical especially if greater particles like prills have to be solidified, which may lead to deformation or sintered lipid blocks in the product container due to an inappropriate time of flight and an incomplete recrystallization. Table VIII gives an overview of some formulation characteristics obtained with spray congealing.
An important point to consider while working with the spray congealing technique is the polymorphic state of the obtained microspheres. As rapid cooling is applied, thermodynamically unstable forms crystallize during the resolidification step. Even microspheres with carnauba wax, which is often stated to be a non-polymorphic material (40,46,221), showed thermal transition events in a microcalorimetric system during storage. However, the effects on parameters such as the dissolution rate were not evaluated (362). A clear polymorphic change was detected with tripalmitin-insulin microspheres, which showed the instable α-form after resolidification and transformed into the stable β-form within 28 days of storage (365). Yajima et al. discovered that the transformation of the α-form of the freshly spray congealed glycerol monostearate Eudragit E microspheres to more stable polymorphic forms had a significant impact on the drug release in a mini-column method used for testing taste masking efficiency (202). Li et al. observed that the drug Bupivacaine as well as tristearin crystallize in their unstable form during spray congealing and the transformation into their stable forms during heat-treatment has a significant impact on the fluidity and gelation behavior in an aqueous medium (363). Although Gelucire 50/13 was able to enhance the dissolution rate of Piroxicam in spray congealed microspheres significantly, the morphological structure changed from a smooth surface to a flake-like structure (“blooming”) and the dissolution rate increased during storage (80). Additionally, even the drug showed a remarkable impact on the polymorphic transformation behavior, for instance, paracetamol was able to stabilize the low melting lipid fraction while caffeine was associated with the transformation to more stable phases (74). Moreover, different polymorphic forms can exhibit a different potential to incorporate drugs in a molecular dispersed form, and therefore changes in lipid polymorphism during storage can lead to drug precipitation and changes in dissolution rate (214). But also the crystallinity and polymorphic state of the drug are important, as melting techniques are able to generate an amorphous state of the drug or reduce crystallinity or have an effect on the recrystallized polymorph of the drug (120,364,369). Hence, studies on polymorphism and drug/lipid interactions are crucial for formulation development.
Freeze Pelletization
Freeze pelletization is a variant of the spray congealing process. The molten dispersion is dropped into a column with a cooling liquid with a needle or nozzle (121,249,370). The liquid offers a better heat transfer than the air and the resulting pellets are described as nonporous, spherically shaped and exhibiting a narrow size distribution (370). Nevertheless, the selection of a suitable cooling fluid seems to be a great challenge, as is has to be non-toxic, inert, immiscible and has to have an appropriate viscosity (370).
Pastillation
Pastillation is a method to facilitate the handling with dusty hazardous powders by transforming them into pastilles common in the petro- and agrochemical industry (250). In this process the molten drug/lipid dispersion is dropped with a needle on a cold surface. An important parameter is the contact angle of the solidified drop to the surface, which affects the flowability of the pastilles and can be adjusted by the needle height and geometry as well as with the temperature of the surface (250,371,372). As the up-scaling to continuous manufacturing seems to be rather easy and different lipid-materials should be applicable, this method seems to be promising for the pharmaceutical industry, provided recrystallization behavior and Reynolds number are appropriate (371).
Fusion (Synonyms: Hot Fusion, Melt Fusion, Melt-Mixing) and Melt-Solidification
The term “hot fusion” refers to the preparation of a solid dispersion of a drug in a matrix and therefore can be understood as a kind of melt extrusion but with the use of less sophisticated equipment on a laboratory scale and has often been applied in pre-formulation studies. Hot fusion is performed by melting the lipid excipient and mixing the drug homogenously by using a magnetic stirrer, a high shear mixer or rotor-stator homogenization to generate a solid dispersion or even a solid solution (29,32,85,153,369). The molten mixture can be molded into tablets (68,373–375), filled in capsules (102,139), poured into a cold water bath and stirred to obtain beads (110,376,377), screened through a sieve for generating granules (32,378), spheronized to pellets (49), or ground and sieved (48,114,136,153,172). The sieved material may be used for direct compression to tablets (32,48,114,136,153,158,169,172,379). The fusion of highly soluble drugs with hydrophobic matrices (e.g., Compritol 888 ATO, glyceryl monostearate, stearic acid, Precirol ATO 5, hydrogenated castor oil etc.) retards the drug release (29,48,114,136,251) and tableting of this solid dispersion has a significantly higher efficiency to sustain release than the direct compression of the physical mixture (29,136,153,210,379). The dissolution of poorly water soluble drugs may be significantly enhanced in a solid dispersion with hydrophilic lipid excipients, such as Gelucire 50/13 (85), Gelucire 44/14 (89,93) compared to the physical mixture of the same (85,93). The addition of an effervescent formulation to the tablets can also help overcome the slower release of tablets compared to an according multiparticulate system (89).
Sintering of Tablets
Thermal treatment (“sintering”) of tablets received by direct compression of the physical mixtures had a significant retarding effect on the dissolution rate due to a redistribution of the wax and an increased matrix tortuosity (158,380,381). However, with hydrogenated cotton seed oil the opposite effect has been seen, which was assumed to be a consequence of wax migration (379). As the study lacks solid-state analysis data a change in polymorphic and morphological properties cannot be excluded (379).
Conclusion
Solvent-free melting techniques are well-known in the literature and very promising for the pharmaceutical industry. The achievable formulation properties serve a wide-area, ranging from modified release by allowing different dissolution kinetics, but also bioavailability enhancement (e.g., gastroretentive formulation), taste masking, up to moisture protection and improvement of swallowability. A recent trend is the design of multiparticulate drug delivery systems, which strike a good and balanced compromise between taste masking and fast release. These systems play a key role in the development of population driven patient-centric strategies, improving the adherence to medication for swallowing difficulties in pediatrics and geriatrics (382). An ongoing challenge in working with lipid materials is the proper handling of stability issues such as polymorphic and morphological changes. Meeting this challenge is worth the effort: the materials required are often cheaper than the corresponding polymers and are mostly processable in standard industrial equipment such as the high shear mixer or fluid bed coater. This facilitates the decision for feasibility studies utilizing a solvent-free melting process in formulation development. Therefore, solvent-free melting processes offer an effective, simple, safe and eco-friendly way of manufacturing in pharmaceutical and food industries.
Abbreviations
- API:
-
Active pharmaceutical ingredient
- BET:
-
Brunauer–Emmett–Teller theory
- Caco:
-
Colon adenocarcinoma cells
- DSC:
-
Differential scanning calorimetry
- DVS:
-
Dynamic vapor sorption
- EDX:
-
Energy dispersive x-ray microanalysis
- FDA:
-
Food and drug administration
- FT-IR:
-
Fourier transform infrared spectroscopy
- GRAS:
-
Generally recognized as safe
- HLB:
-
Hydrophilic lipophilic balance
- HSM:
-
Hot stage polarization microscopy
- IIG:
-
Inactive ingredient guide
- INF:
-
Interferon
- NIR:
-
Near infrared spectroscopy
- PAT:
-
Process analytical technology
- PEG:
-
Polyethylene glycol
- PgP:
-
P-glycoprotein
- SEM:
-
Scanning electron microscope
- TGA:
-
Thermogravimetric analysis
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray diffraction
References
Schwartz JB, Simonelli AP, Higuchi WI. Drug release from wax matrices I. Analysis of data with first-order kinetics and with the diffusion-controlled model. J Pharm Sci. 1968;57(2):274–7.
Lantz RJ, Robinson MJ. Method of preparing sustained release pellets and products thereof. US3146167 A; 1964.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–48.
Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems - an overview. Acta Pharm Sin B. 2013;3(6):361–72.
Hauss DJ. Oral lipid-based formulations. Adv Drug Deliv Rev. 2007;59(7):667–76.
Sohi H, Sultana Y, Khar RK. Taste masking technologies in oral pharmaceuticals: recent developments and approaches. Drug Dev Ind Pharm. 2004;30(5):429–48.
Dubey R, Shami TC, Bhasker Rao KU. Microencapsulation technology and applications. Def Sci J. 2009;59(1):82–95.
Singh MN, Hemant KSY, Ram M, Shivakumar HG. Microencapsulation: a promising technique for controlled drug delivery. Res Pharm Sci. 2010;5(2):65–77.
Jannin V, Musakhanian J, Marchaud D. Approaches for the development of solid and semi-solid lipid-based formulations. Adv Drug Deliv Rev. 2008;60(6):734–46.
Freitas C, Müller RH. Spray-drying of solid lipid nanoparticles (SLN TM). Eur J Pharm Biopharm. 1998;46(2):145–51.
Bagaria SC, Lordi NG. Aqueous dispersions of waxes and lipids for pharmaceutical coating. US5023108 A; 1991.
Chauhan B, Shimpi S, Paradkar A. Preparation and characterization of etoricoxib solid dispersions using lipid carriers by spray drying technique. AAPS PharmSciTech. 2005;6(3):E405–9.
Bose S, Bogner RH. Solventless pharmaceutical coating processes: a review. Pharm Dev Technol. 2007;12(2):115–31.
Grove M, Müllertz A. Liquid self-microemulsifying drug delivery systems. In: Hauss DJ, editor. Oral lipid-based formulations: enhancing the bioavailability of poorly water-soluble drugs. Boca Raton: CRC Press Tayler & Francis Group, LLC; 2007. p. 107-128.
Müller RH, Lippacher A, Gohla S. Solid lipid nanoparticles (SLN) as a carrier system for the controlled release of drugs. In: Wise DL, editor. Handbook of pharmaceutical controlled release technology. New York: Marcel Dekker Inc; 2000. p. 377-392.
Walsh J, Cram A, Woertz K, Breitkreutz J, Winzenburg G, Turner R, et al. Playing hide and seek with poorly tasting paediatric medicines: do not forget the excipients. Adv Drug Deliv Rev. 2014;73:14–33.
Shrestha H, Bala R, Arora S. Lipid-based drug delivery systems. J Pharm. 2014;2014. doi:10.1155/2014/801820.
Cannon JB, Shi Y, Gupta P. Emulsions, microemulsions, and lipid-based drug delivery systems for drug solubilization and delivery - part II: oral applications. In: Liu R, editor. Water-insoluble drug formulation. Boca Raton: CRC Press Tayler & Francis Group, LLC; 2008. p. 195-226.
Wasan KM. Formulation and physiological and biopharmaceutical issues in the development of oral lipid-based drug delivery systems. Drug Dev Ind Pharm. 2001;27(4):267–76.
Sachs-Barrable K, Thamboo A, Lee SD, Wasan KM. Lipid excipients peceol and gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within caco-2 cells. J Pharm Pharm Sci. 2007;10(3):319–31.
Ingelman-Sundberg M, Sim SC, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496–526.
Griffin WC. Classification of surface-active agents by “HLB”. J Soc Cosmet Chem. 1949;1(5):311–26.
Kanicky JR, Lopez-Montilla J-C, Pandey S, Shah DO. Surface chemistry in the petroleum industry. In: Holmberg K, editor. Handbook of applied surface and colloid chemistry. New York: Wiley; 2001. p. 251-267.
Shah MH, Paradkar A. Effect of HLB of additives on the properties and drug release from the glyceryl monooleate matrices. Eur J Pharm Biopharm. 2007;67(1):166–74.
Laitinen R. Physical modification of drug release controlling structures - hydrophobic matrices and fast dissolving particles. Ph.D. Thesis; University of Kuopio, Kuopio, 2009.
Rosiaux Y, Jannin V, Hughes S, Marchaud D. Solid lipid excipients — matrix agents for sustained drug delivery. J Control Release. 2014;188:18–30.
Benameur H, Barthelemy P. Method for coating solid particles with a thermofusible agent, and resulting coated solid particles. US2003/0170312 A1; 2003.
Sinchaipanid N, Junyaprasert V, Mitrevej A. Application of hot-melt coating for controlled release of propranolol hydrochloride pellets. Powder Technol. 2004;141(3):203–9.
Abd El-Halim SM, Amin MM, El-Gazayerly ON, Abd El-Gawad NA. Comparative study on the different techniques for the preparation of sustained-release hydrophobic matrices of a highly water-soluble drug. Drug Discov Ther. 2010;4(6):484–92.
Chandrikapure PL, Wadher KJ, Umekar MJ. Hot-melt coating techniques in sustained release formulation and evaluation of water soluble drug. Int J Pharma Bio Sci. 2011;2(1):273–82.
Ganesh B, Vaishali K, Vishvanath B, Kanchan M, Ashwini C. Formulation and evaluation of sustained release tablet of diltiazem hydrochloride by melt granulation technology. Int Res J Pharm. 2013;4(7):131–7.
Özyazıcı M, Gökçe EH, Ertan G. Release and diffusional modeling of metronidazole lipid matrices. Eur J Pharm Biopharm. 2006;63(3):331–9.
Patil A, Chafle S, Khobragade D, Umathe S, Avari J. Evaluation of hot melt coating as taste masking tool. Int Res J Pharm. 2011;2(8):169–72.
Sudke SG, Sakarakar DM. Design and characterization of modified release pellets of metoprolol tartrate using hot-melt coating excipients. Der Pharm Lett. 2013;5(3):223–9.
Nargis M, Islam MS, Naushin F, Haider SS. Development of sustained release preparations of metoclopramide hydrochloride based on fatty matrix. Dhaka Univ J Pharm Sci. 2012;11(2):129–36.
Balducci AG, Colombo G, Corace G, Cavallari C, Rodriguez L, Buttini F, et al. Layered lipid microcapsules for mesalazine delayed-release in children. Int J Pharm. 2011;421(2):293–300.
Le HT. Preparing sustained release dosage forms of nifedipine by hot-melt coating method. Master Thesis; Oregon State University, Corvallis, 2004.
Uhumwangho MU, Okor RS. Effect of matrix granulation and wax coating on the dissolution rates of paracetamol granules. Afr J Biotechnol. 2006;5(9):766–9.
Uhumwangho MU, Okor RS. Modification of drug release from acetaminophen granules by melt granulation technique - consideration of release kinetics. Pak J Pharm Sci. 2006;19(1):22–7.
Ziegler I, Jacobs I. Coated pellets. US8147874 B2; 2008.
Sudke SG, Sakarkar DM. Application of hot-melt coating for sustained release pellets of fenoverine. Der Pharm Sin. 2013;4(2):153–9.
Breitkreutz J, El-Saleh F, Kiera C, Kleinebudde P, Wiedey W. Pediatric drug formulations of sodium benzoate: II. Coated granules with a lipophilic binder. Eur J Pharm Biopharm. 2003;56(2):255–60.
Krause J, Thommes M, Breitkreutz J. Immediate release pellets with lipid binders obtained by solvent-free cold extrusion. Eur J Pharm Biopharm. 2009;71(1):138–44.
Reitz C, Strachan C, Kleinebudde P. Solid lipid extrudates as sustained-release matrices: the effect of surface structure on drug release properties. Eur J Pharm Sci. 2008;35(4):335–43.
Sax G, Winter G. Mechanistic studies on the release of lysozyme from twin-screw extruded lipid implants. J Control Release. 2012;163(2):187–94.
Johnson WM, Reo JP. Tastemasked pharmaceutical system. US5891476 A; 1999.
Mogna G, Strozzi GP, Mogna L. Food product comprising probiotic bacteria coated with a coating of vegetable origin. EP2544547 A1; 2011.
Abdelkader H, Abdalla OY, Salem H. Formulation of controlled-release baclofen matrix tablets II: influence of some hydrophobic excipients on the release rate and in vitro evaluation. AAPS PharmSciTech. 2008;9(2):675–83.
Biswal S, Sahoo J, Murthy PN, Giradkar RP, Das S, Avari JG. Production variables affecting characteristics of pellets in melt pelletization with wax combination in a laboratory scale spheronizer. Acta Pharm. 2009;59(2):199–210.
Kowalski J, Kalb O, Joshi YM, Serajuddin ATM. Application of melt granulation technology to enhance stability of a moisture sensitive immediate-release drug product. Int J Pharm. 2009;381(1):56–61.
Maejima T, Osawa T, Nakajima K, Kobayashi M. Application of tumbling melt granulation (TMG) method for preparing controlled release beads coated with hydrogenated castor oil. Chem Pharm Bull. 1997;45(5):904–10.
Padsalgi A, Bidkar S, Jadhav V, Sheladiya D. Sustained release tablet of theophylline by hot melt wax coating technology. Asian J Pharm. 2008;2(1):26–9.
Patel JU. Formulation and evaluation of ranolazine sustained release tablets by hot melt coating technique. Ph.D. Thesis; Rajiv Gandhi University of Health Sciences, Karnataka, 2010.
Priyanka J, Bari MM, Barhate SD, Amit SS. Formulation and evaluation of tramadol hydrochloride extended release tablet by using hydrophilic and hydrophobic polymers. Int J Pharm Ind Res. 2013;3(3):243–9.
Thies R, Kleinebudde P. Melt pelletization of a hygroscopic drug in a high shear mixer. Part 3. Effects of binder variation. Chem Pharm Bull. 2001;49(2):140–6.
Tiwari SB, Murthy TK, Pai MR, Mehta PR, Chowdary PB. Controlled release formulation of tramadol hydrochloride using hydrophilic and hydrophobic matrix system. AAPS PharmSciTech. 2003;4(3):Article 31.
Wadher KJ, Kakde RB, Umekar MJ. Formulations of sustained release metformin hydrochloride tablet using combination of lipophilic waxes by melt granulation technique. Afr J Pharm Pharmacol. 2010;4(8):555–61.
Wadher KJ, Kakde RB, Umekar MJ. Sustained release metformin hydrochloride tablet using hydrogenated castor oil and stearic acid by melt granulation technique. Der Pharm Lett. 2010;2(2):64–73.
Maejima T, Osawa T, Nakajima K, Kobayashi M. Preparation of spherical beads without any use of solvents by a novel tumbling melt granulation (TMG) method. Chem Pharm Bull. 1997;45(3):518–24.
Buehler J, Chauhan S, France G. Cimetidine granules coated with a partially hydrogenated vegetable oil. US5597844 A; 1997.
Heng PWS, Wong TW, Cheong WS. Investigation of melt agglomeration process with a hydrophobic binder in combination with sucrose stearate. Eur J Pharm Sci. 2003;19(5):381–93.
Jozwiakowski MJ, Jones DM, Franz RM. Characterization of a hot-melt fluid bed coating process for fine granules. Pharm Res. 1990;7(11):1119–26.
Bequette RJ, Bonenberger BA, Gallian CE, Reckelhoff JR. Direct compression cholestyramine tablet and solvent-free coating therefore. US5455047 A; 1995.
Chansanroj K, Betz G. Improving stability of triglycerides in hot melt coating formulations. Glatt Int Times. 2008;26:1-6.
Chansanroj K, Betz G, Leuenberger H, Mitrevej A, Sinchaipanid N. Development of a multi-unit floating drug delivery system by hot melt coating technique with drug-lipid dispersion. J Drug Delivery Sci Technol. 2007;17(5):333–8.
Chansanroj K, Betz G, Leuenberger H, Mitrevej A, Sinchaipanid N. Polymorphic change of a triglyceride base in hot melt coating process and stability acceleration by tempering process. J Drug Delivery Sci Technol. 2007;17(5):347–52.
Kakiguchi Y, Miyawaki M, Yokota K. Process for producing coated preparation and its use. US6485742 B1; 2003.
Kreye F, Siepmann F, Zimmer A, Willart JF, Descamps M, Siepmann J. Controlled release implants based on cast lipid blends. Eur J Pharm Sci. 2011;43(1-2):78–83.
Montousse C, Pruvost M, Rodriguez F, Brossard C. Extrusion-spheronization manufacture of gelucire (R) matrix beads. Drug Dev Ind Pharm. 1999;25(1):75–80.
Sutananta W, Craig DQM, Newton JM. An investigation into the effects of preparation conditions and storage on the rate of drug-release from pharmaceutical glyceride bases. J Pharm Pharmacol. 1995;47(5):355–9.
Bukovec P, Kroselj V, Turk S, Vrecer F. Optimization of melt pelletization in a high shear mixer. Int J Pharm. 2009;381(2):192–8.
Cavallari C, Rodriguez L, Albertini B, Passerini N, Rosetti F, Fini A. Thermal and fractal analysis microparticles obtained by of diclofenac/gelucire 50/13 ultrasound-assisted atomization. J Pharm Sci. 2005;94(5):1124–34.
Ilic I, Dreu R, Burjak M, Homar M, Kerc J, Srcic S. Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int J Pharm. 2009;381(2):176–83.
Khan N, Craig DQM. The influence of drug incorporation on the structure and release properties of solid dispersions in lipid matrices. J Control Release. 2003;93(3):355–68.
Kroselj V, Pisek R, Vrecer F. Production and characterization of immediate release lansoprazole pellets produced by melt pelletization. Pharm Ind. 2008;70(1):147–55.
Martins RM, Siqueira S, Machado MO, Freitas LA. The effect of homogenization method on the properties of carbamazepine microparticles prepared by spray congealing. J Microencapsul. 2013;30(7):692–700.
Passerini N, Albertini B, Perissutti B, Rodriguez L. Evaluation of melt granulation and ultrasonic spray congealing as techniques to enhance the dissolution of praziquantel. Int J Pharm. 2006;318(1-2):92–102.
Passerini N, Perissutti B, Moneghini M, Voinovich D, Albertini B, Cavallari C, et al. Characterization of carbamazepine-gelucire 50/13 microparticles prepared by a spray-congealing process using ultrasounds. J Pharm Sci. 2002;91(3):699–707.
Pongjanyakul T, Medlicott NJ, Tucker IG. Melted glyceryl palmitostearate (GPS) pellets for protein delivery. Int J Pharm. 2004;271(1-2):53–62.
Qi S, Marchaud D, Craig DQM. An investigation into the mechanism of dissolution rate enhancement of poorly water-soluble drugs from spray chilled gelucire 50/13 microspheres. J Pharm Sci. 2010;99(1):262–74.
Seo A, Holm P, Kristensen HG, Schaefer T. The preparation of agglomerates containing solid dispersions of diazepam by melt agglomeration in a high shear mixer. Int J Pharm. 2003;259(1-2):161–71.
Shimpi SL, Chauhan B, Mahadik KR, Paradkar A. Stabilization and improved in vivo performance of amorphous etoricoxib using gelucire 50/13. Pharm Res. 2005;22(10):1727–34.
Upadhyay P, Pandit JK. Formulation of fast-release gastroretentive solid dispersion of glibenclamide with gelucire 50/13. Trop J Pharm Res. 2012;11(3):361–9.
Vilhelmsen T, Eliasen H, Schaefer T. Effect of a melt agglomeration process on agglomerates containing solid dispersions. Int J Pharm. 2005;303(1-2):132–42.
Vippagunta SR, Maul KA, Tallavajhala S, Grant DJW. Solid-state characterization of nifedipine solid dispersions. Int J Pharm. 2002;236(1-2):111–23.
Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm. 2007;65(1):26–38.
Barker SA, Yap SP, Yuen KH, McCoy CP, Murphy JR, Craig DQ. An investigation into the structure and bioavailability of alpha-tocopherol dispersions in gelucire 44/14. J Control Release. 2003;91(3):477–88.
Barnwell SG, Higginbottom S, Whelan IP, Burns SJ. Pharmaceutical formulations. US6613353 B1; 2003.
Da Fonseca Antunes AB, De Geest BG, Vervaet C, Remon JP. Gelucire 44/14 based immediate release formulations for poorly water-soluble drugs. Drug Dev Ind Pharm. 2013;39(5):791–8.
Damian F, Blaton N, Naesens L, Balzarini J, Kinget R, Augustijns P, et al. Physicochemical characterization of solid dispersions of the antiviral agent UC-781 with polyethylene glycol 6000 and gelucire 44/14. Eur J Pharm Sci. 2000;10(4):311–22.
El Massik MA, Abdallah OY, Galal S, Daabis NA. Semisolid matrix filled capsules: an approach to improve dissolution stability of phenytoin sodium formulation. Drug Dev Ind Pharm. 2003;29(5):531–43.
Hülsmann S, Backensfeld T, Keitel S, Bodmeier R. Melt extrusion – an alternative method for enhancing the dissolution rate of 17β-estradiol hemihydrate. Eur J Pharm Biopharm. 2000;49(3):237–42.
Jatwani S, Rana AC, Singh G, Aggarwal G. Solubility and dissolution enhancement of simvastatin using synergistic effect of hydrophilic carriers. Der Pharm Lett. 2011;3(6):280–93.
Karataş A, Yüksel N, Baykara T. Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol. Farmaco. 2005;60(9):777–82.
Khoo SM, Porter CJH, Charman WN. The formulation of halofantrine as either non-solubilising PEG 6000 or solubilising lipid based solid dispersions: physical stability and absolute bioavailability assessment. Int J Pharm. 2000;205(1-2):65–78.
Parmar N, Bagda A, Patel M, Patel S. Formulation strategy for dissolution enhancement of simvastatin. Int J Pharm Sci Res. 2012;3(10):3817–22.
Siripuram PK, Bandari S, Jukanti R, Veerareddy PR. Formulation and characterization of floating gelucire matrices of metoprolol succinate. Dissolution Technol. 2010;17(3):34–9.
Soliman MS, Khan MA. Preparation and in vitro characterization of a semi-solid dispersion of flurbiprofen with gelucire 44/14 and labrasol. Pharmazie. 2005;60(4):288–93.
Yang D, Kulkarni R, Behme RJ, Kotiyan PN. Effect of the melt granulation technique on the dissolution characteristics of griseofulvin. Int J Pharm. 2007;329(1-2):72–80.
Yuksel N, Karatas A, Ozkan Y, Savaser A, Ozkan SA, Baykara T. Enhanced bioavailability of piroxicam using gelucire 44/14 and labrasol: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2003;56(3):453–9.
Zaky AA, Abdel-Raheem IT. Solubility enhancement of meloxicam prepared via binary and ternary phases using spray congealing. Asian J Pharm Health Sci. 2011;1(4):196–203.
Chauhan B, Shimpi S, Mahadik K, Paradkar A. Preparation and evaluation of floating risedronate sodium gelucire 39/01 matrices. Acta Pharm. 2004;54(3):205–14.
Juarez-Soberanez D, Villafuerte-Robles L. Gelucire 39/01 as excipient for gastroretentive metronidazole sustained delivery. Int J Pharm Pharm Sci. 2011;3(2):86–91.
Shah SH, Patel JK, Patel NV. Floating multiple unit lipid granules of gatifloxacin-gelucire 39/01: formulation optimization using factorial design. Asian J Pharm Sci. 2010;5(1):35–43.
Patel DM, Patel NM, Patel VF, Bhatt DA. Floating granules of ranitidine hydrochloride-gelucire 43/01: formulation optimization using factorial design. AAPS PharmSciTech. 2007;8(2):Article 30.
Rao MEB, Swain SS, Patra CN, Sruti J, Patra S. Development and in vitro evaluation of floating multiparticulate system of repaglinide. FABAD J Pharm Sci. 2011;36(2):75–92.
Shimpi S, Chauhan B, Mahadik KR, Paradkar A. Preparation and evaluation of diltiazem hydrochloride-gelucire 43/01 floating granules prepared by melt granulation. AAPS PharmSciTech. 2004;5(3):51–6.
Charro D, Rial L, Vila Jato JL. Aging of sustained-release formulations of amoxicillin and gelucire 64/02. Drug Dev Ind Pharm. 1993;19(4):473–82.
Yan X, He H, Meng J, Zhang C, Hong M, Tang X. Preparation of lipid aspirin sustained-release pellets by solvent-free extrusion/spheronization and an investigation of their stability. Drug Dev Ind Pharm. 2012;38(10):1221–9.
Kamble R, Maheshwari M, Paradkar A, Kadam S. Melt solidification technique: incorporation of higher wax content in ibuprofen beads. AAPS PharmSciTech. 2004;5(4):75–83.
Patil AT, Khobragade DS, Chafle SA, Ujjainkar AP, Umathe SN, Lakhotia CL. Development and evaluation of a hot-melt coating technique for enteric coating. Braz J Pharm Sci. 2012;48(1):69–77.
Qi S, Deutsch D, Craig DQM. An investigation into the interaction between taste masking fatty acid microspheres and alkaline buffer using thermal and spectroscopic analysis. J Pharm Sci. 2006;95(5):1022–8.
Sudke SG, Sakarakar DM. Design and characterization of enteric coated pellets of aspirin using hot-melt coating technique. Int J Pharma Res Rev. 2013;2(3):1–10.
Bhagwat DA, Kawtikwar PS, Sakarkar DM. Sustained release matrices of verapamil HCl using glyceryl monostearate and stearic acid. Res J Pharm Technol. 2008;1(4):405–9.
Chen H, Shi S, Liu A, Tang X. Combined application of extrusion-spheronization and hot-melt coating technologies for improving moisture-proofing of herbal extracts. J Pharm Sci. 2010;99(5):2444–54.
Kulah G, Kaya O. Investigation and scale-up of hot-melt coating of pharmaceuticals in fluidized beds. Powder Technol. 2011;208(1):175–84.
Robson HJ, Craig DQM, Deutsch D. An investigation into the release of cefuroxime axetil from taste-masked stearic acid microspheres. Part 1: the influence of the dissolution medium on the drug release profile and the physical integrity of the microspheres. Int J Pharm. 1999;190(2):183–92.
Voinovich D, Moneghini M, Perissutti B, Filipovic-Grcic J, Grabnar I. Preparation in high-shear mixer of sustained-release pellets by melt pelletisation. Int J Pharm. 2000;203(1-2):235–44.
Sudke SG, Sakarkar DM. Application of hot-melt coating for sustained release of ofloxacin pellets. Indo Am J Pharm Res. 2013;3(4):3008–14.
Vervaeck A, Saerens L, De Geest BG, De Beer T, Carleer R, Adriaensens P, et al. Prilling of fatty acids as a continuous process for the development of controlled release multiparticulate dosage forms. Eur J Pharm Biopharm. 2013;85(3 Pt A):587–96.
Cheboyina S, Wyandt CM. Wax-based sustained release matrix pellets prepared by a novel freeze pelletization technique II. In vitro drug release studies and release mechanisms. Int J Pharm. 2008;359(1-2):167–73.
El-Shanawany S. Sustained-release of nitrofurantoin from inert wax matrices. J Control Release. 1993;26(1):11–9.
Kennedy JP, Niebergall PJ. Evaluation of extended-release applications for solid dispersion hot-melt fluid bed coatings utilizing hydrophobic coating agents. Pharm Dev Technol. 1998;3(1):95–101.
Kidokoro M, Haramiishi Y, Sagasaki S, Shimizu T, Yamamoto Y. Application of Fluidized Hot-Melt Granulation (FHMG) for the preparation of granules for tableting; properties of granules and tablets prepared by FHMG. Drug Dev Ind Pharm. 2002;28(1):67–76.
Kristensen H, Schaefer T, Thomsen LJ, Kristensen A. Process for the preparation of sustained release pellets. US5807583 A; 1998.
Kukec S, Dreu R, Vrbanec T, Srcic S, Vrecer F. Characterization of agglomerated carvedilol by hot-melt processes in a fluid bed and high shear granulator. Int J Pharm. 2012;430(1-2):74–85.
Nitanai Y, Agata Y, Iwao Y, Itai S. A novel mathematical model considering change of diffusion coefficient for predicting dissolution behavior of acetaminophen from wax matrix dosage form. Int J Pharm. 2012;428(1-2):82–90.
Roblegg E, Jager E, Hodzic A, Koscher G, Mohr S, Zimmer A, et al. Development of sustained-release lipophilic calcium stearate pellets via hot melt extrusion. Eur J Pharm Biopharm. 2011;79(3):635–45.
Thies R, Kleinebudde P. Melt pelletisation of a hygroscopic drug in a high shear mixer Part 1. Influence of process variables. Int J Pharm. 1999;188(2):131–43.
Thies R, Kleinebudde P. Melt pelletisation of a hygroscopic drug in a high shear mixer Part 2. Mutual compensation of influence variables. Eur J Pharm Sci. 2000;10(2):103–10.
Windbergs M, Strachan CJ, Kleinebudde P. Influence of the composition of glycerides on the solid-state behaviour and the dissolution profiles of solid lipid extrudates. Int J Pharm. 2009;381(2):184–91.
Yajima T, Nogata A, Demachi M, Umeki N, Itai S, Yunoki N, et al. Particle design for taste-masking using a spray-congealing technique. Chem Pharm Bull. 1996;44(1):187–91.
Kumar MK, Shah MH, Ketkar A, Mahadik KR, Paradkar A. Effect of drug solubility and different excipients on floating behaviour and release from glyceryl monooleate matrices. Int J Pharm. 2004;272(1-2):151–60.
Sallam AS, Khalil E, Ibrahim H, Freij I. Formulation of an oral dosage form utilizing the properties of cubic liquid crystalline phases of glyceryl monooleate. Eur J Pharm Biopharm. 2002;53(3):343–52.
Brubach JB, Jannin V, Mahler B, Bourgaux C, Lessieur P, Roy P, et al. Structural and thermal characterization of glyceryl behenate by X-ray diffraction coupled to differential calorimetry and infrared spectroscopy. Int J Pharm. 2007;336(2):248–56.
Deore RK, Kavitha K, Tamizhmani TG. Preparation and evaluation of sustained release matrix tablets of tramadol hydrochloride using glyceryl palmitostearate. Trop J Pharm Res. 2010;9(3):275–81.
Hamdani J, Moës AJ, Amighi K. Physical and thermal characterisation of Precirol® and Compritol® as lipophilic glycerides used for the preparation of controlled-release matrix pellets. Int J Pharm. 2003;260(1):47-57.
Hamdani J, Moës AJ, Amighi K. Development and evaluation of prolonged release pellets obtained by the melt pelletization process. Int J Pharm. 2002;245(1-2):167–77.
Jannin V, Pochard E, Chambin O. Influence of poloxamers on the dissolution performance and stability of controlled-release formulations containing Precirol® ATO 5. Int J Pharm. 2006;309(1-2):6–15.
Rao M, Ranpise A, Borate S, Thanki K. Mechanistic evaluation of the effect of sintering on Compritol® 888 ATO matrices. AAPS PharmSciTech. 2009;10(2):355–60.
Reitz C, Kleinebudde P. Influence of thermal and thermo-mechanical treatment. J Therm Anal Calorim. 2007;89(3):669–73.
Reitz C, Kleinebudde P. Solid lipid extrusion of sustained release dosage forms. Eur J Pharm Biopharm. 2007;67(2):440–8.
Barthelemy P, Laforet JP, Farah N, Joachim J. Compritol® 888 ATO: an innovative hot-melt coating agent for prolonged-release drug formulations. Eur J Pharm Biopharm. 1999;47(1):87–90.
Faham A, Prinderre P, Farah N, Eichler KD, Kalantzis G, Joachim J. Hot-melt coating technology. I. Influence of Compritol 888 ATO and granule size on theophylline release. Drug Dev Ind Pharm. 2000;26(2):167–76.
Faham A, Prinderre P, Piccerelle P, Farah N, Joachim J. Hot melt coating technology: influence of Compritol 888 ATO and granule size on chloroquine release. Pharmazie. 2000;55(6):444–8.
Farah N, Barthelemy P, Joachim J. Method for preparing a pharmaceutical composition with modified release of the active principle, comprising a, matrix. US6194005 B1; 2001.
Griffin EN, Niebergall PJ. Release kinetics of a controlled-release multiparticulate dosage form prepared using a hot-melt fluid bed coating method. Pharm Dev Technol. 1999;4(1):117–24.
Hamdani J, Goole J, Moës AJ, Amighi K. In vitro and in vivo evaluation of floating riboflavin pellets developed using the melt pelletization process. Int J Pharm. 2006;323(1-2):86–92.
Hamdani J, Moës AJ, Amighi K. Development and in vitro evaluation of a novel floating multiple unit dosage form obtained by melt pelletization. Int J Pharm. 2006;322(1-2):96–103.
Jannin V, Berard V, Andres C. Modification of the drug release of ibuprofen by hot-melt coating with mixes of Compritol™ 888 ATO and non-ionic surfactants. AAPS. 2005;7(S1):Article -0853.
Kavitha K, Deore RK, Tamizhmani TG. Preparation and evaluation of sustained release matrix tablets of tramadol hydrochloride using Compritol 888 ATO by melt granulation technique. Res J Pharm Biol Chem Sci. 2010;1(3):431–40.
Knezevic Z, Gosak D, Hraste M, Rausl D, Khan MZI. Application of hot-melt coating process for designing a lipid based controlled release drug delivery system for highly aqueous soluble drugs. Chem Pharm Bull. 2009;57(5):464–71.
Li FQ, Hu JH, Deng JX, Su H, Xu S, Liu JY. In vitro controlled release of sodium ferulate from Compritol 888 ATO-based matrix tablets. Int J Pharm. 2006;324(2):152–7.
Molke M, Iqbal M, Rao KS. Formulation and evaluation of verapamil HCl gastroretentive floating tablet from matrices prepared using Compritol ATO 888. Res J Pharm Biol Chem Sci. 2010;1(3):422–30.
Obaidat AA, Obaidat RM. Controlled release of tramadol hydrochloride from matrices prepared using glyceryl behenate. Eur J Pharm Biopharm. 2001;52(2):231–5.
Parab PV, Oh CK, Ritschel WA. Sustained release from Precirol® (glycerol palmito-stearate) matrix. Effect of mannitol and hydroxypropyl methylcellulose on the release of theophylline. Drug Dev Ind Pharm. 1986;12(8-9):1309–27.
Pivette P, Faivre V, Mancini L, Gueutin C, Daste G, Ollivon M, et al. Controlled release of a highly hydrophilic API from lipid microspheres obtained by prilling: Analysis of drug and water diffusion processes with X-ray-based methods. J Control Release. 2012;158(3):393–402.
Zhang YE, Schwartz JB. Melt granulation and heat treatment for wax matrix-controlled drug release. Drug Dev Ind Pharm. 2003;29(2):131–8.
Windbergs M, Strachan CJ, Kleinebudde P. Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur J Pharm Biopharm. 2009;71(1):80–7.
Windbergs M, Strachan CJ, Kleinebudde P. Investigating the principles of recrystallization from glyceride melts. AAPS PharmSciTech. 2009;10(4):1224–33.
Windbergs M, Strachan CJ, Kleinebudde P. Tailor-made dissolution profiles by extruded matrices based on lipid polyethylene glycol mixtures. J Control Release. 2009;137(3):211–6.
Becker K, Haack D, Salar-Behzadi S, Zimmer A. Oral pharmaceutical composition comprising taste-masked N-acetylcysteine. EP13163638.3; 2013.
Haupt M, Thommes M, Heidenreich A, Breitkreutz J. Lipid-based intravesical drug delivery systems with controlled release of trospium chloride for the urinary bladder. J Control Release. 2013;170(2):161–6.
Mayama H. Blooming theory of tristearin. Soft Matter. 2009;5(4):856–9.
Sax G, Feil F, Schulze S, Jung C, Brauchle C, Winter G. Release pathways of interferon alpha 2a molecules from lipid twin screw extrudates revealed by single molecule fluorescence microscopy. J Control Release. 2012;162(2):295–302.
Sakarkar DM, Dorle AK, Mahajan NM, Sudke SG. Design of sustained release pellets of ferrous fumarate using cow ghee as hot-melt coating agent. Int J Pharm Investig. 2013;3(3):151–6.
Sakarkar DM, Jaiswal SB, Dorle AK, Deshmukh VN. Application of cow ghee as hot melt coating agent in the design of sustained release pellets. Int J PharmTech Res. 2009;1(4):1167–72.
Akiyama Y, Yoshioka M, Horibe H, Hirai S, Kitamori N, Toguchi H. pH-independent controlled-release microspheres using polyglycerol esters of fatty-acids. J Pharm Sci. 1994;83(11):1600–7.
Abdelbary G, Prinderre P, Eouani C, Joachim J, Reynier JP, Piccerelle P. The preparation of orally disintegrating tablets using a hydrophilic waxy binder. Int J Pharm. 2004;278(2):423–33.
Lang P, Szuts A, Ambrus R, Szabóné RP. The applicability of sucrose laurate in hot-melt technology. Acta Pharm Hung. 2011;81(1):43–6.
Szuts A, Láng P, Ambrus R, Kiss L, Deli MA, Szabó-Révész P. Applicability of sucrose laurate as surfactant in solid dispersions prepared by melt technology. Int J Pharm. 2011;410(1-2):107–10.
Abd-Elbary A, Tadros MI, Alaa-Eldin AA. Sucrose stearate-enriched lipid matrix tablets of etodolac: modulation of drug release, diffusional modeling and structure elucidation studies. AAPS PharmSciTech. 2013;14(2):656–68.
Szuts A. Study of the applicability of sucrose esters in hot-melt technology. Ph.D. Thesis; University of Szeged, Szeged, 2009.
Lutton ES. Review of the polymorphism of saturated even glycerides. J Am Oil Chem Soc. 1950;27(7):276–81.
Malkin T. The polymorphism of glycerides. Prog Chem Fats Other Lipids. 1954;2:1–50.
Chapman D. The polymorphism of glycerides. Chem Rev. 1962;62(5):433–56.
Sato K. Crystallization behaviour of fats and lipids—a review. Chem Eng Sci. 2001;56(7):2255–65.
Ghotra BS, Dyal SD, Narine SS. Lipid shortenings: a review. Food Res Int. 2002;35(10):1015–48.
Himawan C, Starov VM, Stapley AGF. Thermodynamic and kinetic aspects of fat crystallization. Adv Colloid Interface Sci. 2006;122(1-3):3–33.
Fang W, Mayama H, Tsujii K. Spontaneous formation of fractal structures on triglyceride surfaces with reference to their super water-repellent properties. J Phys Chem B. 2007;111(3):564–71.
Hong BY, Li D, Lei QF, Xu L, Fang WJ, Mayama H. Kinetics on formation of super water repellent surfaces from phase transformation in binary mixtures of trimyristin and tripalmitin. Colloids Surf A Physicochem Eng Asp. 2012;396:130–6.
Witzleb R, Mullertz A, Kanikanti VR, Hamann HJ, Kleinebudde P. Dissolution of solid lipid extrudates in biorelevant media. Int J Pharm. 2012;422(1-2):116–24.
Khan N, Craig DQM. Role of blooming in determining the storage stability of lipid-based dosage forms. J Pharm Sci. 2004;93(12):2962–71.
Roussin P, Duddu S. Semi-solid matrix of polyglycolized glycerides and nucleation enhancer. US6171615 B1; 2001.
Amrutkar PP, Patil SB, Todarwal AN, Wagh MA, Kothawade PD, Surawase RK. Design and evaluation of taste masked chewable dispersible tablet of lamotrigine by melt granulation. Int J Drug Deliv. 2010;2(2):183–91.
Griffin WC. Calculation of HLB values of non-ionic surfactants. J Soc Cosmet Chem. 1954;5(4):249–56.
Grassi M, Voinovich D, Grabnar I, Franceschinis E, Perissutti B, Filipovic-Grcic J. Preparation and in vitro/in vivo characterisation of a melt pelletised paracetamol/stearic acid sustained release delivery system. Spectrosc Int J. 2004;18(2):375–86.
Grassi M, Voinovich D, Moneghini M, Franceschinis E, Perissutti B, Filipovic-Grcic J. Preparation and evaluation of a melt pelletised paracetamol/stearic acid sustained release delivery system. J Control Release. 2003;88(3):381–91.
Bakala N’Goma JC, Amara S, Dridi K, Jannin V, Carriere F. Understanding the lipid-digestion processes in the GI tract before designing lipid-based drug-delivery systems. Ther Deliv. 2012;3(1):105–24.
Ljusberg-Wahren H, Nielsen FS, Brogard M, Troedsson E, Mullertz A. Enzymatic characterization of lipid-based drug delivery systems. Int J Pharm. 2005;298(2):328–32.
Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14(2):115–22.
Christensen JØ, Schultz K, Mollgaard B, Kristensen HG, Mullertz A. Solubilisation of poorly water-soluble drugs during in vitro lipolysis of medium- and long-chain triacylglycerols. Eur J Pharm Sci. 2004;23(3):287–96.
Christiansen A, Backensfeld T, Weitschies W. Effects of non-ionic surfactants on in vitro triglyceride digestion and their susceptibility to digestion by pancreatic enzymes. Eur J Pharm Sci. 2010;41(2):376–82.
Olbrich C, Kayser O, Müller RH. Lipase degradation of Dynasan 114 and 116 solid lipid nanoparticles (SLN) - effect of surfactants, storage time and crystallinity. Int J Pharm. 2002;237(1-2):119–28.
Mohsin K. Design of lipid-based formulations for oral administration of poorly water-soluble drug fenofibrate: effects of digestion. AAPS PharmSciTech. 2012;13(2):637–46.
Michalk A, Kanikanti VR, Hamann HJ, Kleinebudde P. Controlled release of active as a consequence of the die diameter in solid lipid extrusion. J Control Release. 2008;132(1):35–41.
Witzleb R, Kanikanti VR, Hamann HJ, Kleinebudde P. Solid lipid extrusion with small die diameters - electrostatic charging, taste masking and continuous production. Eur J Pharm Biopharm. 2011;77(1):170–7.
Masic I, Ilic I, Dreu R, Ibric S, Parojcic J, Duric Z. An investigation into the effect of formulation variables and process parameters on characteristics of granules obtained by in situ fluidized hot melt granulation. Int J Pharm. 2012;423(2):202–12.
Suzuki H, Onishi H, Hisamatsu S, Masuda K, Takahashi Y, Iwata M, et al. Acetaminophen-containing chewable tablets with suppressed bitterness and improved oral feeling. Int J Pharm. 2004;278(1):51–61.
Suzuki H, Onishi H, Takahashi Y, Iwata M, Machida Y. Development of oral acetaminophen chewable tablets with inhibited bitter taste. Int J Pharm. 2003;251(1-2):123–32.
Gittings S, Turnbull N, Roberts CJ, Gershkovich P. Dissolution methodology for taste masked oral dosage forms. J Control Release. 2014;173:32–42.
Yajima T, Itai S, Takeuchi H, Kawashima Y. Optimum heat treatment conditions for masking the bitterness of the clarithromycin wax matrix. Chem Pharm Bull. 2003;51(11):1223–6.
Woertz K, Tissen C, Kleinebudde P, Breitkreutz J. Development of a taste-masked generic ibuprofen suspension: top-down approach guided by electronic tongue measurements. J Pharm Sci. 2011;100(10):4460-70.
Monteagudo E, Langenheim M, Salerno C, Buontempo F, Bregni C, Carlucci A. Pharmaceutical optimization of lipid-based dosage forms for the improvement of taste-masking, chemical stability and solubilizing capacity of phenobarbital. Drug Dev Ind Pharm. 2014;40(6):783–92.
Sato K, Ueno S. Physical properties of fats in food. In: Rajah K, editor. Fats in food technology. Chichester: Wiley; 2014. p. 1–38.
Marangoni AG, Wesdorp LH. Structure and properties of fat crystal networks. Boca Raton: CRC Press Taylor & Francis Group. 2013.
Schulze S, Winter G. Lipid extrudates as novel sustained release systems for pharmaceutical proteins. J Control Release. 2009;134(3):177–85.
Windbergs M, Gueres S, Strachan CJ, Kleinebudde P. Two-step solid lipid extrusion as a process to modify dissolution behavior. AAPS PharmSciTech. 2010;11(1):2–8.
Reitz C, Kleinebudde P. Spheronization of solid lipid extrudates. Powder Technol. 2009;189(2):238–44.
Omar M. Studying the release of diclofenac sodium from glycerides. Int J Pharm Pharm Sci. 2013;5(4):119–27.
Smith KW, Bhaggan K, Talbot G, Malssen KF. Crystallization of fats: influence of minor components and additives. J Am Oil Chem Soc. 2011;88(8):1085–101.
Ribeiro APB, Masuchi MH, Miyasaki EK, Domingues MAF, Stroppa VLZ, de Oliveira GM, et al. Crystallization modifiers in lipid systems. J Food Sci Technol. 2014. doi:10.1007/s13197-014-1587-0.
Lopes DG, Becker K, Faulhammer E, Haack D, Lochmann D, Khinast JG, et al. Advanced structuring of lipids for controlled drug release; polymorphic crystallizations and curing effects. Atlanta: 14 AIChE Annual Meeting; 2014.
Xia D, Cui F, Gan Y, Mu H, Yang M. Design of lipid matrix particles for fenofibrate: effect of polymorphism of glycerol monostearate on drug incorporation and release. J Pharm Sci. 2014;103(2):697–705.
Cerdeira M, Martini S, Candal RJ, Herrera ML. Polymorphism and growth behavior of low-trans fat blends formulated with and without emulsifiers. J Am Oil Chem Soc. 2006;83(6):489–96.
Herrera ML, Marquez Rocha FJ. Effects of sucrose ester on the kinetics of polymorphic transition in hydrogenated sunflower oil. J Am Oil Chem Soc. 1996;73(3):321–6.
Oh J-H, McCurdy AR, Clark S, Swanson BG. Stabilizing polymorphic transitions of tristearin using diacylglycerols and sucrose polyesters. J Am Oil Chem Soc. 2005;82(1):13–9.
Aronhime J, Sarig S, Garti N. Dynamic control of polymorphic transformation in triglycerides by surfactants: the button syndrome. J Am Oil Chem Soc. 1988;65(7):1144–50.
Garti N, Wellner E, Sarig S. Crystal structure modifications of tristearin by food emulsifiers. J Am Oil Chem Soc. 1982;59(4):181–5.
Aronhime JS, Sarig S, Garti N. Mechanistic considerations of polymorphic transformations of tristearin in the presence of emulsifiers. J Am Oil Chem Soc. 1987;64(4):529–33.
Gowan GW, Bruce RD. Aliphatic esters as a solventless coating for pharmaceuticals. CA 2082137; 1992.
Yoshioka M, Horibe H, Kashihara T. Granulated preparations and method of producing the same. US5443846 A; 1995.
Holstborg J, Pedersen B, Krog N, Olesen S. Physical properties of diglycerol esters in relation to rheology and stability of protein-stabilised emulsions. Colloids Surf B Biointerfaces. 1999;12(3-6):383–90.
Thomas A, Müller SS, Frey H. Beyond poly(ethylene glycol): linear polyglycerol as a multifunctional polyether for biomedical and pharmaceutical applications. Biomacromolecules. 2014;15(6):1935–54.
Sutananta W, Craig DQM, Newton JM. An investigation into the effect of preparation conditions on the structure and mechanical-properties of pharmaceutical glyceride bases. Int J Pharm. 1994;110(1):75–91.
Pivette P, Faivre V, Brubach JB, Daste G, Ollivon M, Lesieur S. Polymorphism of glyceryl behenates: from the individual compounds to the pharmaceutical excipient. Chem Phys Lipids. 2014;183:191–203.
Long C, Zhang L, Qian Y. Preparation and crystal modification of ibuprofen-loaded solid lipid microparticles. Chin J Chem Eng. 2006;14(4):518–25.
Patel JK, Patel NV, Shah SH. In vitro controlled release of colon targeted mesalamine from Compritol ATO 888 based matrix tablets using factorial design. Res Pharm Sci. 2009;4(2):63–75.
Bhoop BS. Quality by Design (QbD) for holistic pharma excellence and regulatory compliance. Pharm Times. 2014;46(8):26–33.
Guidance for industry—Q8(R2) pharmaceutical development. Food and Drug Administration; 2009: p. 1-25.
Guidance for industry—Q3A impurities in new drug substances. Food and Drug Administration; 2008: p. 1–14.
World Health Organization. Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. World Health Organ Tech Rep Ser. 2009;953:87–130.
Breitkreutz J, Matilainen J. Lipid pellets with enhanced taste-masking. US20110262549 A1; 2011.
El-Refaie W, El-Massik M, Abdallah O, Khalafallah N. Formulation and evaluation of taste-masked paracetamol-lipid sachets and chewable tablets. J Pharm Investig. 2014;44(6):431–42.
Vaassen J, Bartscher K, Breitkreutz J. Taste masked lipid pellets with enhanced release of hydrophobic active ingredient. Int J Pharm. 2012;429(1-2):99–103.
Lakkis JM. Encapsulation and controlled release in backery applications. In: Lakkis JM, editor. Encapsulation and controlled release technologies in food systems. Ames: Blackwell Publishing Inc; 2007. p. 113-134.
Mehuys E, Vervaet C, Remon JP. Hot-melt extruded ethylcellulose cylinders containing a HPMC-Gelucire® core for sustained drug delivery. J Control Release. 2004;94(2-3):273–80.
Shiino K, Iwao Y, Miyagishima A, Itai S. Optimization of a novel wax matrix system using aminoalkyl methacrylate copolymer E and ethylcellulose to suppress the bitter taste of acetaminophen. Int J Pharm. 2010;395(1-2):71–7.
Dordunoo SK, Ford JL, Rubinstein MH. Solidification studies of polyethylene glycols, gelucire 44/14 or their dispersions with triamterene or temazepam. J Pharm Pharmacol. 1996;48(8):782–9.
Bodmeier R, Paeratakul O, Chen H, Zhang W. Formation of sustained release wax matrices within hard gelatin capsules in a fluidized bed. Drug Dev Ind Pharm. 1990;16(9):1505–19.
Galal S, El Massik MA, Abdallah OY, Daabis NA. Study of in-vitro release characteristics of carbamazepine extended release semisolid matrix filled capsules based on gelucires. Drug Dev Ind Pharm. 2004;30(8):817–29.
Vial-Bernasconi AC, Buri P, Doelker E, Beyssac E, Duchaix G, Aiache JM. In vivo evaluation of an indomethacin monolithic, extended zero-order release hard-gelatin capsule formulation based on saturated polyglycolysed glycerides. Pharm Acta Helv. 1995;70(4):307–13.
Wu PC, Tsai MJ, Huang YB, Chang JS, Tsai YH. In vitro and in vivo evaluation of potassium chloride sustained release formulation prepared with saturated polyglycolyed glycerides matrices. Int J Pharm. 2002;243(1-2):119–24.
Adnan S, Nisar-Ur-Rahman, Ahmed S, Khan SM, Saeed-Ul-Hassar S. In vitro comparison of sustained release hydroxypropylmethyl cellulose and lipid-based matrix systems of diltiazem HCl. Pak J Zool. 2010;42(6):515–9.
San Vicente A, Hernandez RM, Gascon AR, Calvo MB, Pedraz JL. Effect of aging on the release of salbutamol sulfate from lipid matrices. Int J Pharm. 2000;208(1-2):13–21.
Dennis AB, Farr SJ, Kellaway IW, Taylor G, Davidson R. In vivo evaluation of rapid release and sustained release gelucire capsule formulations. Int J Pharm. 1990;65(1-2):85–100.
Keen JM, McGinity JW, Williams III RO. Enhancing bioavailability through thermal processing. Int J Pharm. 2013;450(1-2):185–96.
Quinten T, De Beer T, Vervaet C, Remon JP. Evaluation of injection moulding as a pharmaceutical technology to produce matrix tablets. Eur J Pharm Biopharm. 2009;71(1):145–54.
Cheboyina S, Wyandt CM. Wax-based sustained release matrix pellets prepared by a novel freeze pelletization technique - I. Formulation and process variables affecting pellet characteristics. Int J Pharm. 2008;359(1-2):158–66.
Shukla D, Chakraborty S, Singh S, Mishra B. Pastillation: a novel technology for development of oral lipid based multiparticulate controlled release formulation. Powder Technol. 2011;209(1-3):65–72.
Kato Y, Sunada H, Yonezawa Y, Ishino R. Sustained-release mechanisms of wax matrix system for controlled-release. Chem Pharm Bull. 1994;42(8):1646–50.
Srivastava S, Mishra G. Fluid bed technology : overview and parameters for process selection. Int J Pharm Sci Drug Res. 2010;2(4):236–46.
Jannin V, Cuppok Y. Hot-melt coating with lipid excipients. Int J Pharm. 2013;457(2):480–7.
Barbosa-Canovas GV, Ortega-Rivas E, Pablo J, Yan H. Food powders: physical properties, processing, and functionality. New York: Kluwer Academic/Plenum Publishers; 2005.
Walker G, Bell SEJ, Vann M, Jones DS, Andrews G. Fluidised bed characterisation using Raman spectroscopy: applications to pharmaceutical processing. Chem Eng Sci. 2007;62(14):3832–8.
Bogomolov A, Engler M, Melichar M, Wigmore A. In-line analysis of a fluid bed pellet coating process using a combination of near infrared and Raman spectroscopy. J Chemom. 2010;24(7-8):544–57.
Lee MJ, Seo DY, Lee HE, Wang IC, Kim WS, Jeong MY, et al. In line NIR quantification of film thickness on pharmaceutical pellets during a fluid bed coating process. Int J Pharm. 2011;403(1-2):66–72.
Kukec S, Hudovornik G, Dreu R, Vrecer F. Study of granule growth kinetics during in situ fluid bed melt granulation using in-line FBRM and SFT probes. Drug Dev Ind Pharm. 2014;40(7):952–9.
Penhasi A. Composition and method for improving stability and extending shelf life of probiotic bacteria and food products thereof. EP2648528 A1; 2013.
Maronga S. On the optimization of the fluidized bed particulate coating process. Ph.D. Thesis; Royal Institute of Technology, Stockholm, 1998.
Kukec S, Vrecer F, Dreu R. A study of in situ fluid bed melt granulation using response surface methodology. Acta Pharm. 2012;62(4):497–513.
Deepak S, Dinesh K, Mankaran S, Gurmeet S, Singh RM. Taste masking technologies: a novel approach for the improvement of organoleptic property of pharmaceutical active substance. Int Res J Pharm. 2012;3(4):108–16.
Sudke SG, Sakarakar DM. Hot-melt coating: an innovative pharmaceutical coating technique. J Pharm Res Clin Pract. 2013;3(1):16–26.
Sudke SG, Sakarakar DM. Lipids—an instrumental excipient in pharmaceutical hot-melt coating. Int J PharmTech Res. 2013;5(2):607–21.
Koller DM, Hannesschläger G, Leitner M, Khinast JG. Non-destructive analysis of tablet coatings with optical coherence tomography. Eur J Pharm Sci. 2011;44(1-2):142–8.
Wirges M, Muller J, Kasa P, Regdon G, Pintye-Hodi K, Knop K, et al. From mini to micro scale-feasibility of Raman spectroscopy as a process analytical tool (PAT). Pharmaceutics. 2011;3(4):723–30.
Gendre C, Genty M, Boiret M, Julien M, Meunier L, Lecoq O, et al. Development of a process analytical technology (PAT) for in-line monitoring of film thickness and mass of coating materials during a pan coating operation. Eur J Pharm Sci. 2011;43(4):244–50.
Chavda VP, Soniwala MM, Chavda JR. Particle coating: from conventional to advanced. Int J Pharm Med al Res. 2013;1:1–17.
Rantanen J, Wikstrom H, Turner R, Taylor LS. Use of in-line near-infrared spectroscopy in combination with chemometrics for improved understanding of pharmaceutical processes. Anal Chem. 2005;77(2):556–63.
Wikstrom H, Marsac PJ, Taylor LS. In-line monitoring of hydrate formation during wet granulation using Raman spectroscopy. J Pharm Sci. 2005;94(1):209–19.
Litster J, Ennis B. The science and engineering of granulation processes. Heidelberg: Springer Science & Business Media; 2004.
Bauman I, Ćurić D, Boban M. Mixing of solids in different mixing devices. Sadhana. 2008;33(6):721–31.
Campisi B, Vojnovic D, Chicco D, Phan-Tan-Luu R. Melt granulation in a high shear mixer: optimization of mixture and process variables using a combined experimental design. Chemom Intell Lab Syst. 1999;48(1):59–70.
Voinovich D, Moneghini M, Perissutti B, Franceschinis E. Melt pelletization in high shear mixer using a hydrophobic melt binder: influence of some apparatus and process variables. Eur J Pharm Biopharm. 2001;52(3):305–13.
Augsburger LL, Hoag SW, editors. Pharmaceutical dosage forms-tablets. Boca Raton: CRC Press; 2008.
Holm P. High shear mixer granulators. In: Parikh DM, editor. Handbook of pharmaceutical granulation technology. New York: Marcel Dekker; 1997. p. 151-204.
Passerini N, Calogera G, Albertini B, Rodriguez L. Melt granulation of pharmaceutical powders: a comparison of high-shear mixer and fluidised bed processes. Int J Pharm. 2010;391(1-2):177–86.
Kraciuk R, Sznitowska M. Effect of different excipients on the physical characteristics of granules and tablets with carbamazepine prepared with polyethylene glycol 6000 by fluidized hot-melt granulation (FHMG). AAPS PharmSciTech. 2011;12(4):1241–7.
DiNunzio J, Repka MA, Langley N, editors. Melt extrusion: materials, technology and drug product design. New York: Springer; 2013.
Crowley MM, Zhang F, Repka MA, Thumma S, Upadhye SB, Battu SK, et al. Pharmaceutical applications of hot-melt extrusion: part I. Drug Dev Ind Pharm. 2007;33(9):909–26.
Maniruzzaman M, Boateng JS, Snowden MJ, Douroumis D. A review of hot-melt extrusion: process technology to pharmaceutical products. ISRN Pharm. 2012;2012. doi:10.5402/2012/436763.
Muehlenfeld C, Thommes M. Miniaturization in pharmaceutical extrusion technology: feeding as a challenge of downscaling. AAPS PharmSciTech. 2012;13(1):94–100.
Zecevic DE, Wagner KG. Rational development of solid dispersions via hot-melt extrusion using screening, material characterization, and numeric simulation tools. J Pharm Sci. 2013;102(7):2297–310.
Lu M, Guo Z, Li Y, Pang H, Lin L, Liu X, et al. Application of hot melt extrusion for poorly water-soluble drugs: limitations, advances and future prospects. Curr Pharm Des. 2014;20(3):369–87.
Okuro PK, Eustáquio de Matos Jr F, Favaro-Trindade CS. Technological challenges for spray chilling encapsulation of functional food ingredients. Food Technol Biotechnol. 2013;51(2):171–82.
Garti N, McClements DJ. Encapsulation technologies and delivery systems for food ingredients and nutraceuticals. Cambridge: Woodhead Publishing; 2012.
Gouin S. Microencapsulation: industrial appraisal of existing technologies and trends. Trends Food Sci Technol. 2004;15(7-8):330–47.
Maltesen MJ, van de Weert M, Grohganz H. Design of experiments-based monitoring of critical quality attributes for the spray-drying process of insulin by NIR spectroscopy. AAPS PharmSciTech. 2012;13(3):747–55.
Chan LW, Tan LH, Heng PWS. Process analytical technology: application to particle sizing in spray drying. AAPS PharmSciTech. 2008;9(1):259–66.
Chokshi R, Zia H. Hot-melt extrusion technique: a review. Iran J Pharm Res. 2004;3:3–16.
Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54(2):107–17.
Ghali ES, Klinger GH, Schwartz JB. Thermal treatment of beads with wax for controlled release. Drug Dev Ind Pharm. 1989;15(9):1311–28.
Giles HF, Wagner JR, Mount EM. Extrusion the definitive processing guide and handbook. New York: William Andrew Inc.; 2005.
Patel A, Sahu D, Dashora A, Garg R, Agraval P, Patel P, et al. A review of hot melt extrusion technique. Int J Innov Res Sci Eng Technol. 2013;2(6):2194–8.
Repka MA, Shah S, Lu JN, Maddineni S, Morott J, Patwardhan K, et al. Melt extrusion: process to product. Expert Opin Drug Deliv. 2012;9(1):105–25.
Repka MA, Majumdar S, Kumar Battu S, Srirangam R, Upadhye SB. Applications of hot-melt extrusion for drug delivery. Expert Opin Drug Deliv. 2008;5(12):1357–76.
Mehuys E, Vervaet C, Gielen I, Van Bree H, Remon JP. In vitro and in vivo evaluation of a matrix-in-cylinder system for sustained drug delivery. J Control Release. 2004;96(2):261–71.
Mehuys E, Remon JP, Korst A, Van Bortel L, Mols R, Augustijns P, et al. Human bioavailability of propranolol from a matrix-in-cylinder system with a HPMC-Gelucire core. J Control Release. 2005;107(3):523–36.
Nakamichi K, Izumi S, Yasuura H. Method of manufactoring wax matrices. US5700410 A; 1997.
Edimo A, Leterme P, Denis J, Traisnel M, Gayot AT. Capacity of lipophilic auxiliary substances to give spheres by extrusion-spheronization. Drug Dev Ind Pharm. 1993;19(7):827–42.
Miyagawa Y, Okabe T, Yamaguchi Y, Miyajima M, Sato H, Sunada H. Controlled-release of diclofenac sodium from wax matrix granule. Int J Pharm. 1996;138(2):215–24.
Sato H, Miyagawa Y, Okabe T, Miyajima M, Sunada H. Dissolution mechanism of diclofenac sodium from wax matrix granules. J Pharm Sci. 1997;86(8):929–34.
Liu J, Zhang F, McGinity JW. Properties of lipophilic matrix tablets containing phenylpropanolamine hydrochloride prepared by hot-melt extrusion. Eur J Pharm Biopharm. 2001;52(2):181–90.
De Brabander C, Vervaet C, Fiermans L, Remon JP. Matrix mini-tablets based on starch/microcrystalline wax mixtures. Int J Pharm. 2000;199(2):195–203.
Quintavalle U, Voinovich D, Perissutti B, Serdoz F, Grassi G, Dal Col A, et al. Preparation of sustained release co-extrudates by hot-melt extrusion and mathematical modelling of in vitro/in vivo drug release profiles. Eur J Pharm Sci. 2008;33(3):282–93.
Vithani K, Maniruzzaman M, Slipper IJ, Mostafa S, Miolane C, Cuppok Y, et al. Sustained release solid lipid matrices processed by hot-melt extrusion (HME). Colloids Surf B Biointerfaces. 2013;110:403–10.
Oliveira G, Wahl MA, Pinto JF. Characterization of laminar extrudates manufactured at room temperature in the absence of solvents for the delivery of drugs. Int J Pharm. 2013;454(1):90–8.
Madan S, Madan S. Hot melt extrusion and its pharmaceutical applications. Asian J Pharm Sci. 2012;7(2):123–33.
Repka MA, Battu SK, Upadhye S, Thumma S. Pharmaceutical applications of hot-melt extrusion: part II. Drug Dev Ind Pharm. 2007;33(10):1043–57.
Keen JM, Hughey JR, Bennett RC, Jannin V, Rosiaux Y, Marchaud D, et al. Effect of tablet structure on controlled release from supersaturating solid dispersions containing glyceryl behenate. Mol Pharm. 2015;12(1):120–6.
Choy YW, Khan N, Yuen KH. Significance of lipid matrix aging on in vitro release and in vivo bioavailability. Int J Pharm. 2005;299(1-2):55–64.
Norn V, editor. Emulsifiers in food technology. Chichester: Wiley-Blackwell; 2015.
Varzakas T, Tzia C, editors. Food engineering handbook: food process engineering. Boca Raton: CRC Press, 2014.
Akiyama Y, Yoshioka M, Horibe H, Hirai S, Kitamori N, Toguchi H. Novel oral controlled-release microspheres using polyglycerol esters of fatty acids. J Control Release. 1993;26(1):1–10.
Rothrock DA, Cheetham HC. Hot melt coating. US2285095 A; 1942.
NIIR Board of Consultants & Engineers. The complete technology book on wax and polishes. Asia Pacific Business Press Inc. 2011.
Ustunol Z. Edible films and coatings for meat and poultry. In: Embuscado M, Huber KC, editors. Edible Films and coatings for food applications. Heidelberg: Springer Science and Business Media; 2009. p. 245-268.
Dewettinck K, Huyghebaert A. Fluidized bed coating in food technology. Trends Food Sci Technol. 1999;10(4-5):163–8.
Hoy FH. Simulated fat and method of preparing the same. US2290854 A; 1942.
Norton FH. Coating with hot melt c3-c4 polyolefin packaging compositions and articles obtained thereby. US3145111 A; 1964.
Clarke C. The science of ice cream. Cambridge: The Royal Society of Chemistry; 2004.
Schrooyen PMM, van der Meer R, De Kruif CG. Microencapsulation: its application in nutrition. Proc Nutr Soc. 2001;60:475–9.
Rotman A, Blatt Y. Heat stabilized flavoring agents coated with hydrogenated castor oil. US5098725 A; 1992.
Merchant ZM, Behringer MU, Hill LG, McPherson AE, Tangprasertchai, Uraiwan Vogt RA, Nayyar DK. Low temperature cooking cereal-based products. US6599550 B2; 2003.
Hanus EJ, Ouellette PA, Stoyle LE. Chewable fatty coating of iron particles. US3035985 A; 1962.
Hanus EJ, Ouellette PA, Stoyle LE. Chewable, palatable, vitamin b preparations. US3037911 A; 1962.
Tuerck PA, McVean DE. Formula modifications in a solvent-free tablet film coat. J Pharm Sci. 1973;62(9):1534–7.
Tuerek PA. Tablet coating. US3383237 A; 1968.
Jones DM, Percel PJ. Coating of multiparticulates using molten materials - formulation and process considerations. In: Ghebre-Sellassie I, editor. Multiparticulate oral drug delivery. New York: Marcel Dekker, Inc; 1994. p. 113-142.
Patel HL, Patel HB, Davuluri C, Modasiya MK. Review on solventless coating technology. Am J PharmTech Res. 2011;1(4):154–73.
Achanta AS, Adusumilli PS, James KW, Rhodes CT. Development of hot melt coating methods. Drug Dev Ind Pharm. 1997;23(5):441–9.
Choi MMS, Meisen A. Sulfur coating of urea in shallow spouted beds. Chem Eng Sci. 1997;52(7):1073–86.
Paulucci KP, Stabile KR, Freitas LAP. Study of the batch hot melt coating of tablets in spouted bed. Proc 14th Int Dry Symp 2004;A:225–31.
Weiss PJ, Meisen A. Laboratory studies on sulfur-coating urea by the spouted bed process. Can J Chem Eng. 1983;61(3):440–7.
Kennedy JP, Niebergall PJ. Development and optimization of a solid dispersion hot-melt fluid bed coating method. Pharm Dev Technol. 1996;1(1):51–62.
Kraahs P, Bold S, Fahsel L. Melt-coated pharmaceutical composition with fast release. EP2365801 A1; 2011.
Santus G, Golzi R. Prompt-release pharmaceutical compositions. WO1997018798 A1; 1997.
Pham L, Christensen JM. Preparation of acetaminophen capsules containing beads prepared by hot-melt direct blend coating. Pharm Dev Technol. 2014;19(1):91–102.
Ayres JW. Hot melt coating by direct blending and coated substrates. US20070141071 A1; 2007.
Chaudhari PD, Chaudhari SP, Yeola GS, Barhate NS. Melt granulation technique: a review. Latest Rev. 2006;4(1). Available from: http://www.pharmainfo.net/reviews/melt-granulation-technique-review.
Srivastav M, Prabhakar B, Omray A. Extended release tablet technologies—matrix, melt granulation and multiparticulates: an overview. Int J Univ Pharm Life Sci. 2011;1(1):331–54.
Pauli-Bruns A, Knop K, Lippold BC. Preparation of sustained release matrix pellets by melt agglomeration in the fluidized bed: influence of formulation variables and modelling of agglomerate growth. Eur J Pharm Biopharm. 2010;74(3):503–12.
Seo A, Holm P, Schaefer T. Effects of droplet size and type of binder on the agglomerate growth mechanisms by melt agglomeration in a fluidised bed. Eur J Pharm Sci. 2002;16(3):95–105.
Challis D, Heafield J, Knott TJ, Leslie ST, Malkowska STA, Miller RB, et al. Sustained release compositions and a method of preparing pharmaceutical compositions. US6162467 A; 2000.
Schaefer T, Holm P, Kristensen HG. Melt granulation in a laboratory scale high shear mixer. Drug Dev Ind Pharm. 1990;16(8):1249–77.
Schæfer T. Growth mechanisms in melt agglomeration in high shear mixers. Powder Technol. 2001;117(1-2):68–82.
Thomsen LJ, Schaefer T, Kristensen HG. Prolonged release matrix pellets prepared by melt pelletization II. Hydrophobic substances as meltable binders. Drug Dev Ind Pharm. 1994;20(7):1179–97.
Thomsen LJ, Schæfer T, Sonnergaard JM, Kristensen HG. Prolonged release matrix pellets prepared by melt pelletization I. Process variables. Drug Dev Ind Pharm. 1993;19(15):1867–87.
Eliasen H, Schaefer T, Kristensen HG. Effects of binder rheology on melt agglomeration in a high shear mixer. Int J Pharm. 1998;176(1):73–83.
Eliasen H, Kristensen HG, Schaefer T. Electrostatic charging during a melt agglomeration process. Int J Pharm. 1999;184(1):85–96.
Iveson SM, Litster JD, Hapgood K, Ennis BJ. Nucleation, growth and breakage phenomena in agitated wet granulation processes: a review. Powder Technol. 2001;117(1-2):3–39.
Vervaet C, Remon P. Melt granulation. In: Parikh DM, editor. Handbook of pharmaceutical granulation technology. Boca Raton: CRC Press; 2009. p. 435-448
Shiino K, Iwao Y, Fujinami Y, Itai S. Preparation and evaluation of granules with pH-dependent release by melt granulation. Int J Pharm. 2012;431(1-2):70–7.
Mašić I, Ilić I, Dreu R, Ibrić S, Parojčić J, Srčič S. Melt granulation in fluidized bed: a comparative study of spray-on versus in situ procedure. Drug Dev Ind Pharm. 2014;40(1):23–32.
Aleksić I, Duriš J, Ilić I, Ibrić S, Parojčić J, Srčič S. In silico modeling of in situ fluidized bed melt granulation. Int J Pharm. 2014;466(1-2):21–30.
Prado HJ, Bonelli PR, Cukierman AL. In situ fluidized hot melt granulation using a novel meltable binder: effect of formulation variables on granule characteristics and controlled release tablets. Powder Technol. 2014;264:498–506.
Voinovich D, Campisi B, Moneghini M, Vincenzi C, Phan-Tan-Luu R. Screening of high shear mixer melt granulation process variables using an asymmetrical factorial design. Int J Pharm. 1999;190(1):73–81.
Walker GM, Holland CR, Ahmad MMN, Craig DQM. Influence of process parameters on fluidised hot-melt granulation and tablet pressing of pharmaceutical powders. Chem Eng Sci. 2005;60(14):3867–77.
Jannin V, Berard V, N’Diaye A, Andres C, Pourcelot Y. Comparative study of the lubricant performance of Compritol® 888 ATO either used by blending or by hot melt coating. Int J Pharm. 2003;262(1-2):39–45.
Halle PD, Sakhare RS, Dadage KK, Birajdar GO, Raut DB. A review on melt granulation technique. J Pharm Phyther. 2013;1(3):6–10.
Pivette P, Faivre V, Daste G, Ollivon M, Lesieur S. Rapid cooling of lipid in a prilling tower. J Therm Anal Calorim. 2009;98(1):47–55.
Emås M, Nyqvist H. Methods of studying aging and stabilization of spray-congealed solid dispersions with carnauba wax. 1. Microcalorimetric investigation. Int J Pharm. 2000;197(1-2):117–27.
Li LC, Zhu L, Song J-F, Deng J-S, Bandopadhyay R, Wurster DE. Effect of solid state transition on the physical stability of suspensions containing bupivacaine lipid microparticles. Pharm Dev Technol. 2005;10(2):309–18.
Savolainen M, Herder J, Khoo C, Lovqvist K, Dahlqvist C, Glad H, et al. Evaluation of polar lipid-hydrophilic polymer microparticles. Int J Pharm. 2003;262(1-2):47–62.
Maschke A, Becker C, Eyrich D, Kiermaier J, Blunk T, Gopferich A. Development of a spray congealing process for the preparation of insulin-loaded lipid microparticles and characterization thereof. Eur J Pharm Biopharm. 2007;65(2):175–87.
Passerini N, Perissutti B, Albertini B, Voinovich D, Moneghini M, Rodriguez L. Controlled release of verapamil hydrochloride from waxy microparticles prepared by spray congealing. J Control Release. 2003;88(2):263–75.
Albertini B, Passerini N, Gonzalez-Rodriguez ML, Perissutti B, Rodriguez L. Effect of Aerosil on the properties of lipid controlled release microparticles. J Control Release. 2004;100(2):233–46.
Rodriguez L, Passerini N, Cavallari C, Cini M, Sancin P, Fini A. Description and preliminary evaluation of a new ultrasonic atomizer for spray-congealing processes. Int J Pharm. 1999;183(2):133–43.
Serajuddin AT. Solid dispersion of poorly water-soluble drugs: early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–66.
Cheboyina S, Chambliss WG, Wyandt CM. A novel freeze pelletization technique for preparing matrix pellets. Pharm Technol. 2004;28(10):98–110.
Kim JW, Ulrich J. Prediction of degree of deformation and crystallization time of molten droplets in pastillation process. Int J Pharm. 2003;257(1-2):205–15.
Shukla D, Chakraborty S, Singh S, Mishra B. Lipid-based oral multiparticulate formulations—advantages, technological advances and industrial applications. Expert Opin Drug Deliv. 2011;8(2):207–24.
Chime SA, Attama AA, Kenechukwu FC, Umeyor EC, Onunkwo GC. Formulation, in vitro and in vivo characterisation of diclofenac potassium sustained release tablets based on solidified reverse micellar solution (SRMS). Br J Pharm Res. 2013;3(1):90–107.
Kreye F, Siepmann F, Zimmer A, Willart JF, Descamps M, Siepmann J. Cast lipid implants for controlled drug delivery: importance of the tempering conditions. J Pharm Sci. 2011;100(8):3471–81.
Siepmann J, Siepmann F. Mathematical modeling of drug release from lipid dosage forms. Int J Pharm. 2011;418(1):42–53.
Paradkar AR, Maheshwari M, Ketkar AR, Chauhan B. Preparation and evaluation of ibuprofen beads by melt solidification technique. Int J Pharm. 2003;255(1-2):33–42.
Maheshwari M, Ketkar AR, Chauhan B, Patil VB, Paradkar AR. Preparation and characterization of ibuprofen-cetyl alcohol beads by melt solidification technique: effect of variables. Int J Pharm. 2003;261(1-2):57–67.
Uhumwangho MU, Ramana Murthy KV. Release characteristics of diltiazem hydrochloride wax-matrix granules-thermal sintering effect. J Appl Sci Environ Manag. 2011;15(2):365–70.
Sudha BS, Sridhar BK, Srinatha A. Modulation of tramadol release from a hydrophobic matrix: implications of formulations and processing variables. AAPS PharmSciTech. 2010;11(1):433–40.
Zhang YE, Tchao R, Schwartz JB. Effect of processing methods and heat treatment on the formation of wax matrix tablets for sustained drug release. Pharm Dev Technol. 2001;6(2):131–44.
Rao MRP, Ranpise AA, Thanki KC, Borate SG, Parikh GN. Effect of processing and sintering on controlled release wax matrix tablets of ketorolac tromethamine. Indian J Pharm Sci. 2009;71(5):538–44.
Wahlich J, Stegemann S, Orlu-Gul M. Meeting commentary-“medicines for older adults: learning from practice to develop patient centric drug products”. Int J Pharm. 2013;456(1):251–7.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Becker, K., Salar-Behzadi, S. & Zimmer, A. Solvent-Free Melting Techniques for the Preparation of Lipid-Based Solid Oral Formulations. Pharm Res 32, 1519–1545 (2015). https://doi.org/10.1007/s11095-015-1661-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-015-1661-y