
Abstract
Solid tumors arising from malignant transformation of glial cells are one of the leading causes of central nervous system tumor-related death in children. Recurrence in spite of rigorous surgical and chemoradiation therapies remains a major hurdle in management of these tumors. Here, we investigate the efficacy of the second-generation receptor tyrosine kinase inhibitor nilotinib as a therapeutic option for the management of pediatric gliomas. We have utilized two independent pediatric high-grade glioma cell lines with either high platelet-derived growth factor receptor alpha (PDGFRα) or high PDGFRβ expression in in vitro assays to investigate the specific downstream effects of nilotinib treatment. Using in vitro cell-based assays we show that nilotinib inhibits PDGF-BB-dependent activation of PDGFRα. We further show that nilotinib is able to decrease cell proliferation and anchorage-independent growth via suppression of AKT and ERK1/2 signaling pathways. Our results suggest that nilotinib may be effective for management of a PDGFRα-dependent group of pediatric gliomas.
Similar content being viewed by others


Introduction
Malignant glial neoplasms comprise 8–10 % of primary pediatric central nervous system (CNS) tumors [1], and are the most common cause of solid tumor-related death in children [2]. These lesions are classified as grade III or IV by the World Health Organization (WHO), and histology is most commonly anaplastic astrocytoma (AA; grade III) and glioblastoma (GBM; grade IV) [3]. The cornerstones of therapy remain surgical resection and adjuvant chemoradiation, but tumor recurrence is inevitable and long-term response to treatment remains extremely poor. Development of novel therapies has been restricted by limited understanding of tumor biology and dependence upon preclinical models based on adult disease.
Genomic studies of pediatric high-grade gliomas (HGG) have demonstrated that the most frequent copy number aberration (CNA) is focal amplification of the platelet-derived growth factor receptor (PDGFRA) locus at chromosome 4q12, encoding the α isoform of platelet-derived growth factor receptor (PDGFRα) [4–7]. Several exome sequencing profiles of high-grade pediatric supratentorial and brainstem gliomas revealed a frequent, novel mutation of H3F3A, encoding histone variant H3.3, that is associated with poorer overall survival [8, 9]; among brainstem lesions, amplification of PDGFRA occurred exclusively in H3F3A-mutant tumors [10]. PDGFRα protein expression corresponds to gene amplification, and is also seen in some tumors that do not contain PDGFRA amplification [11]. Expression of PDGFR correlates with high-grade histology in pediatric gliomas [12], and associated activation of downstream Ras- and AKT-pathway signaling mediators has been correlated with poor survival [13, 14].
The second-generation receptor tyrosine kinase inhibitor nilotinib (Tasigna; Novartis Pharmaceuticals) binds its targets with similar affinity to the prototype molecule imatinib (Gleevec; Novartis Pharmaceuticals) [15–19]. Unlike imatinib, it is not dependent upon the OCT-1 transporter for cell influx [20], and although it may be subject to transporter-dependent efflux [21, 22], it is detectable within the brains of mice with an intact blood–brain barrier [23]. Currently in use for treating imatinib-resistant and newly diagnosed chronic myeloid leukemia, nilotinib has a well-established safety and toxicity profile [24, 25]. In this study, we examine the effects of nilotinib on pediatric GBM-derived cell lines, in order to understand the biochemical and biological impact of inhibiting PDGFR signaling and to evaluate its suitability as a therapeutic target.
Materials and methods
Cell lines and culture
Pediatric GBM cell lines SJ-G2 and SF-188 [26, 27] were a gift from Dr. Nada Jabado (McGill University, Montreal, Quebec), and normal human astrocytes (NHA) immortalized with E7 and hTERT were a gift from Dr. Russell Pieper (University of California, San Francisco, CA, USA). Adult GBM cell lines U-118, U-87 and U-251 were obtained from the American Type Culture Collection (ATCC). All cell lines were cultured as adherent monolayers in minimal media supplemented with 10 % fetal bovine serum (FBS), at 37 °C in 5 % CO2.
Evaluation of exogenous ligand stimulation and inhibition
Methods and results are presented below for SJ-G2; parallel experiments carried out in SF-188 are presented in supplementary sections (see detailed methods in Supplement). Culture stimulation was performed using dimerized B-isoform of recombinant human platelet-derived growth factor (PDGF-BB; Cell Signaling Technology), which has affinity for both α and β isoforms of PDGFR. Cell cultures at 70–80 % confluence were incubated overnight in growth media containing 0.5 % FBS, then washed with warm phosphate-buffered saline (PBS), and exposed to fresh minimal media containing PDGF-BB at working concentration. Stimulation was terminated by placing cultures on ice and washing with cold PBS, then scraping and suspending in cell lysis buffer.
Western immunoblot assay
Cell cultures were scraped and lysed on ice in 1X Cell Lysis Buffer (Cell Signaling) containing protease (Roche Diagnostics) and phosphatase inhibitors (Calbiochem). Protein was quantified using bicinchoninic acid (BCA) assay (Thermo Scientific), and lysate containing 25–40 μg protein was loaded onto 7–12.5 % SDS-PAGE gels for electrophoresis. A detailed list of antibodies used is presented in Supplementary methods.
Cell viability, proliferation and colony formation assays
Cell viability was assessed by MTS assay using CellTiter 96® Aqueous One Solution reagent (Promega) according to the manufacturer’s instructions. Direct count of viable cells using the trypan blue dye exclusion method was performed with the Vi-CELL Cell Viability Analyzer (Beckman Coulter). For cell proliferation assay, chemiluminescent Cell Proliferation ELISA (Roche) using 5-bromo-2′-deoxyuridine (BrdU) labeling was carried out according to the manufacturer’s instructions, using a 12-h labeling period. Clonogenic assay and soft-agar colony-forming assay were performed to evaluate in vitro growth potential [28]. Colonies were fixed in 10 % formalin, then stained with 0.05 % crystal violet. Each condition was carried out in triplicate, in three independent experiments. Detailed methods are described in Supplementary methods.
Subcutaneous xenograft
SJ-G2 cells (1 × 106) were injected into the right flank of 6–8-week-old NOD–SCID mice (Jackson Laboratory). On detection of palpable tumor, mice were given nilotinib or vehicle by oral gavage (10 mg/kg daily), and sacrificed upon signs of sickness or at the end of the 10-week study. Excised tumor dimensions were measured with calipers, and volume estimated by the formula 4πabc/3, where a, b and c represent tumor radius in three planes. Animal use protocols were approved by the animal care committed of the Hospital for Sick Children (Toronto, Canada) under AUP#2656.
Results
PDGFR protein expression in pediatric GBM cell lines
To assess relative expression levels of PDGFR proteins, the pediatric GBM cell lines SJ-G2 and SF-188 were compared to adult GBM cell lines and to non-transformed human astrocytes using immunoblotting. We found that NHA expressed PDGFRβ but not PDGFRα. SF-188 followed this pattern, by expressing minimal PDGFRβ and not PDGFRα. In contrast, SJ-G2 contained large amounts of PDGFRα, and far less PDGFRβ (Fig. 1a). Results for SJ-G2 are presented throughout, and parallel experiments carried out in SF-188 are presented in Supplementary Data and Figures. Neither pediatric GBM cell line was found to contain high-level amplification of PDGFRA (Supplementary Fig. 1S). In summary, pediatric GBM—like adult GBM—contains variable expression of the two PDGF receptors.

Expression and activation of PDGFRα. a Variable expression of PDGFRα and β is demonstrated in pediatric (SF-188, SJ-G2) and adult (U-87, U-251, U-118) glioblastoma cell-lines. b Western blot analysis of PDGFRα phosphorylation following exposure to exogenous PDGF-BB under varying conditions. Vinculin is loading control. SJ-G2 cells were exposed to 0–100 ng/mL PDGF-BB for 5 min. A concentration-dependent increase in PDGFRα phosphorylation was observed, to a plateau ≥50 ng/mL. c SJ-G2 cells were exposed to 100 ng/mL PDGF-BB for 0–60 min. Phosphorylation of PDGFRα occurred rapidly, with a peak observed following 1 min of ligand exposure
Activation of PDGFRα is PDGF-BB-dependent
When grown in low-serum conditions, SJ-G2 cells contained minimal phosphorylated PDGFRα. Addition of exogenous PDGF-BB to growth media resulted in phosphorylation of PDGFRα. Phosphorylation increased with increasing ligand concentration to a plateau ≥50 ng/mL (Fig. 1b). Based on this finding, to ensure maximal phosphorylation, we carried out subsequent stimulation using PDGF-BB at a concentration of 80 ng/mL. The largest fraction of phosphorylated receptor was seen following one minute of stimulation, the earliest time point measured (Fig. 1c). As exposure time to ligand was lengthened, the amount of phosphorylated receptor and total receptor decreased. In order to ensure reproducibility during subsequent stimulation, we chose a stimulation duration of 5 min.
PDGFRα stimulation and downstream signaling is inhibited by nilotinib
We observed a dose-dependent effect on number of viable cells in culture between 0 and 3 μM nilotinib (see below), so chose an overlapping concentration range to examine signaling effects. To evaluate the effect of PDGFR activation on the downstream AKT and Ras pathways, we monitored phosphorylation of AKT S473 and ERK1/2 T202/Y204 in the presence and absence of nilotinib. In low-serum growth conditions, background phosphorylation of PDGFRα was minimal, and activation of AKT and ERK1/2 was not detected (Fig. 2a). Exposure to exogenous PDGF-BB resulted in increased phosphorylation of PDGFRα, as well as AKT and ERK1/2. The presence of 3 or 5 μM nilotinib during PDGF-BB stimulation resulted in little PDGFRα activation, and the phosphorylation of AKT and ERK1/2 were also significantly reduced. The activation of PDGFRα was evaluated using an antibody specific to phospho-tyrosine 754, but immunoblotting with a pan-phosphotyrosine antibody also demonstrated that phosphorylation of tyrosine residues at the molecular weight of PDGFR was generally reduced in the presence of nilotinib (Fig. 2b).

Nilotinib inhibits PDGF-BB-dependent activation of PDGFRα. SJ-G2 cultures in low-serum growth conditions were stimulated with exogenous PDGF-BB in the absence and presence of nilotinib. Accompanying bar graphs display relative phosphorylation in each condition, compared with stimulation in 0 μM nilotinib treatment (maximal stimulation). Significant difference is indicated by * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. a Western blot analysis of PDGFRα, AKT and ERK1/2 phosphorylation. β-actin is loading control. Minimal basal phosphorylation of PDGFRα was observed, and little activation of downstream signaling mediators. Exposure to PDGF-BB resulted in phosphorylation of PDGFRα and activation of AKT and ERK1/2. Treatment with nilotinib decreased PDGFRα phosphorylation and the associated activation of downstream signaling mediators. b Immunoblotting of total cell lysate detected global reduction of phospho-tyrosine at the molecular weight of PDGFRα in the presence of nilotinib. Vinculin is loading control
Nilotinib inhibition of PDGFRα in serum decreases activation of AKT and ERK1/2 signaling
We then evaluated the effects of niloinib inhibition in the more complex milieu of serum-supplemented growth media, and additionally examined the phosphorylation of S6 ribosomal protein, which effects enhanced mRNA translation in AKT-stimulated cell growth, and of MEK, a direct activator of ERK1/2. In serum-supplemented growth media, a low basal level of PDGFRα phosphorylation was observed (Fig. 3, row 1 of graphs). Basal activation of AKT, S6 ribosomal protein, MEK and ERK1/2 was observed. To evaluate the effect of nilotinib on activation of PDGFRα and its signaling pathways, each inhibition condition was compared to this basal phosphorylation. Cultures were first exposed to 0–10 μM nilotinib for 2 h (Fig. 3, first column of graphs). With increasing nilotinib concentration, phosphorylation of PDGFRα was significantly decreased. During this time period, a small decrease in AKT activation was noted, as well as a marked decrease in S6 ribosomal protein phosphorylation. (Fig. 3, rows 2 and 3 of graphs). When exposure to 3 μM nilotinib was then extended to 6 and 24 h (Fig. 3, second column of graphs), a further reduction in AKT activation was seen, and the decreased phosphorylation of S6 ribosomal protein was sustained. Initial exposure to nilotinib resulted in stimulation of MEK activation (Fig. 3, row 4 of graphs). As exposure to 3 μM nilotinib was prolonged, however, MEK phosphorylation decreased, and after 24 h, was suppressed below baseline. ERK1/2 activation was not significantly altered during short-duration nilotinib treatment, but decreased following 24 h of exposure (Fig. 3, row 5 of graphs).

Nilotinib inhibits PDGFRα and the AKT and ERK1/2 signaling pathways. SJ-G2 cultures in 10 % FBS-supplemented growth media were exposed to nilotinib 0–10 μM for 2 h, and to 3 μM for up to 24 h. Phosphorylation of PDGFRα, AKT, S6 ribosomal protein, MEK and ERK1/2 was analyzed by Western blotting. β-actin is loading control. Accompanying bar graphs display relative phosphorylation in each condition, compared with basal levels (0 μM nilotinib vehicle treatment). Significant difference is indicated by * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. Basal phosphorylation of all proteins was demonstrated. Exposure to nilotinib resulted in reduction of PDGFRα phosphorylation. Phosphorylation of AKT and S6 ribosomal protein was also decreased, and continually suppressed with extended inhibitor treatment. Phosphorylation of MEK was initially increased, but sustained exposure to nilotinib resulted in suppression below baseline. ERK1/2 activation was decreased following 24 h of nilotinib treatment
Nilotinib decreases SJ-G2 proliferation
A dose-dependent effect on cell viability was demonstrated by MTS assay when SJ-G2 cells were treated with nilotinib (0–10 μM) (Fig. 4a). Following ≥1 day of exposure to the inhibitor, the viability of all treated cultures was less than that of vehicle-treated control. After ≥2 days of exposure, fewer viable cells were seen in cultures exposed to ≥3 μM nilotinib compared to cultures in 1 μM nilotinib. There was no consistent difference in effect on viable cell numbers between 3 and 10 μM nilotinib. To determine whether the effect on viability was sustained, SJ-G2 was cultured in the presence of nilotinib (0–10 μM) up to 14 days (Fig. 4b). Number of viable cells in treated cultures was significantly less than in vehicle-treated control, initially noted after 4 days of inhibition and continued through 14 days of exposure. This result was corroborated using direct counting of viable cells using trypan blue dye exclusion; when treated for 4 or more days, all SJ-G2 cultures exposed to ≥3 μM nilotinib contained significantly fewer viable cells compared to vehicle-treated control (Fig. 4c). There was no consistent difference among the cultures treated with 3–10 μM nilotinib. Apoptosis assays were also performed but no significant differences were noted (data not shown).

Nilotinib reduces SJ-G2 growth and proliferation. Cell cultures treated with nilotinib were compared with vehicle-treated controls. Significant difference is indicated by * for p < 0.05, ** for p < 0.01, and *** for p < 0.001. a SJ-G2 cultures were exposed to nilotinib (0–10 μM) for 1–4 days. MTS assay demonstrated a dose-dependent effect on numbers of viable cells in culture. All treated cultures demonstrated significantly fewer viable cells compared to control, and a further difference was seen between 1 μM and ≥3 μM nilotinib treatment. b Durability of effect on cell numbers was evaluated by exposing SJ-G2 cultures to nilotinib (0–10 μM) for 2–14 days. Treated cultures exhibited fewer viable cells up to 14 days. c SJ-G2 cultures were exposed to nilotinib (0–10 μM) for 2–6 days. The number of viable cells, as determined by exclusion of trypan blue dye, was lower in the presence of nilotinib. d SJ-G2 cultures were exposed to nilotinib (0–10 μM) for 2–4 days. Incorporation of BrdU as detected by ELISA was decreased in treated cultures
To evaluate the effect of nilotinib on SJ-G2 proliferation, we assessed the incorporation of BrdU by chemiluminescent ELISA (Fig. 4d). Following 2 and 4 days of exposure, all treated cultures (3–10 μM) generated significantly less luminescent product than vehicle-treated control.
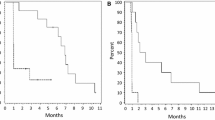
Nilotinib decreases in vitro and in vivo tumorigenicity
The number of colonies formed in clonogenic assay was decreased in the presence of all concentrations of nilotinib (Fig. 5a). Anchorage-independent growth of SJ-G2 cells suspended in soft agar was also significantly decreased by exposure to nilotinib (Fig. 5b). Mice harboring SJ-G2 xenograft tumors survived significantly longer (p = 0.0006) when treated with nilotinib, compared with mice given drug vehicle (Fig. 5c). Tumors excised from treated mice were smaller at the time of animal sacrifice (Fig. 5d). Two treated animals did not become ill; at the end of the study period, the subcutaneous masses that had been identified were found to be fibrofatty tissue.

Nilotinib reduces SJ-G2 in vitro and in vivo growth. a Plate-attached SJ-G2 cells were exposed to nilotinib (0–10 μM) for 21 days. Colony numbers in the presence of nilotinib were compared to vehicle-treated controls; significant difference is indicated by ** for p < 0.01 and *** for p < 0.001. b SJ-G2 cells suspended in 0.35 % agar were exposed to nilotinib (0–10 μM) for 21 days. Colony numbers in the presence of nilotinib were compared to vehicle-treated controls; significant difference is indicated by *** for p < 0.001. c Kaplan–Meier plot for NOD–SCID mice bearing SJ-G2 flank tumors. Animals treated with nilotinib demonstrated prolonged survival. d Excised xenograft tumors that had been exposed to nilotinib were smaller than those from vehicle-treated mice. Significant difference is indicated by * for p < 0.05
Discussion
Demonstration of PDGFRA amplification as the most common CNA among pediatric HGG, and the association of histone H3.3 mutation in malignant pediatric brain-stem glioma with PDGFRA amplification, identified PDGFRα and its signaling pathways as likely integral to the malignant behavior of some tumors. It is therefore identified as a potential therapeutic target. Recognizing that understanding of this disease and advancement of therapy has historically been hindered by reliance upon studies of adult gliomas, we have carried out investigations of PDGFRα signaling and inhibition in cell lines derived from pediatric glioblastoma tumors.
Pediatric high-grade tumors exhibit variability in PDGFRA copy number, as well as in expression of PDGFRα and in platelet-derived growth factor (PDGF) itself [11, 29–31]. Although the SJ-G2 cell line does not contain amplification of PDGFRA, its expression of high levels of PDGFRα, the activation of which is dependent on dosage of ligand (PDGF-BB), suggests that it represents tumors that use PDGFRα signaling to support their tumorigenic behavior. To our knowledge, this is the first demonstration in a pediatric GBM cell line that activation of PDGFR is dependent upon ligand binding. This stands in contrast, for instance, to constitutively-activated mutant forms of epidermal growth factor receptor that are frequently found in adult GBM [32, 33]. Activation of PDGFRα occurred rapidly, with peak phosphorylation seen within a minute of exposure to PDGF-BB, and decayed, suggesting that normal mechanisms of receptor internalization and degradation remain functional in these cells [34].
Ligand-mediated PDGFR stimulation activates the PI3K–AKT and Raf–MEK–ERK signaling pathways [35–37] and is dependent upon cell type and developmental stage [38–40]. Stimulation with PDGF-BB of SJ-G2 cells resulted in phosphorylation of PDGFRα and corresponding activation of AKT and ERK1/2. Exposure to nilotinib inhibited PDGF-BB-mediated stimulation of the receptor, and markedly decreased activation of AKT and ERK1/2. Phosphorylation of tyrosine in response to PDGF-BB was generally decreased in the presence of nilotinib, suggesting that PDGFRα auto-activation was inhibited.
The effect of nilotinib inhibition was subsequently examined in the heterogeneous environment of serum-supplemented growth media. In this setting, we observed a low basal level of PDGFRα phosphorylation. Consistent with ongoing PDGFRα stimulation, we also detected basal activation of AKT, S6 ribosomal protein, MEK and ERK1/2, although activation of other cell-surface receptors may have contributed to this. Exposure to nilotinib decreased PDGFRα phosphorylation in all conditions. Phosphorylation of AKT and S6 ribosomal protein was rapidly reduced, and was maintained at a significantly decreased level during sustained inhibitor exposure, suggesting that some component of their activation is PDGFR-dependent. Interestingly, there was an early increase in MEK phosphorylation, within the first 2 h of nilotinib exposure. The rapidity of this change suggests that it results from an alteration in steady-state balance between signaling pathways.
Since inhibition with nilotinib suppressed PDGFRα-mediated signaling, we then investigated its effect on SJ-G2 cell numbers and proliferation. Our results show that cell proliferation and clonogenic potential (in vitro tumor-forming ability) were significantly reduced at 3 μM nilotinib compared to vehicle-treated control. The reduced proliferation resulted in fewer viable cells over the same time interval of growth compared to untreated controls. As patients receiving typical oral dosaging of nilotinib achieve a mean peak serum concentration of 3.6 μM [24], the plateau in effect suggests that maximal anti-PDGFRα effect occurs at a concentration that can be achieved safely in the clinical context. The in vitro results were supported by in vivo findings of prolonged survival in nilotinib-treated mice bearing SJ-G2 tumors, and smaller tumor sizes in the treated mice.
These functional assays indicate that the result of nilotinib-mediated decrease in PDGFRα activation and signaling is a reduction in SJ-G2 proliferation, colony formation and in vivo growth. This overall decrease in tumorigenic behavior suggests that SJ-G2 may represent tumors that depend upon PDGFRα signaling and would therefore respond to nilotinib inhibition.
In contrast, SF-188 expressed very little PDGFRα and demonstrated high basal activation of AKT and ERK1/2, suggesting that this cell line is dependent upon activation of alternative growth factor receptor pathways to transduce mitogenic signals (Supplementary Figs. 2S, 3S, 4S). Consistent with this, we found that treatment with nilotinib had little effect on activation of AKT and ERK1/2. Furthermore, while treated cultures of SF-188 initially demonstrated lower viability, this difference was lost with sustained inhibition. SF-188 contains a mutation of the tumor suppressor and Ras-GAP NF1 [41], which likely maintains activation of both AKT and ERK1/2 pathways by failing to regulate Ras activation. In contrast to SJ-G2, which contains wildtype NF1, SF-188 can be seen to represent the subset of tumors that do not depend upon PDGFRα activation.
All chemotherapeutic agents intended for use in the CNS must be able to penetrate the blood–brain barrier. Although its predecessor imatinib has poor CSF penetration [42], regarding this question nilotinib remains under clinical investigation. Interestingly, nilotinib demonstrates in vivo activity against Abelson tyrosine kinase (Abl) within neurons of murine basal ganglia [23].
As our understanding of the biology of pediatric HGG evolves, so does our recognition that they are a clinical and pathological entity distinct from adult tumors—and, indeed, that they encompass different subtypes. In this study, we identified the SJ-G2 cell line as representative of tumors that are dependent upon PDGFRα expression and activation to drive signaling pathways leading to tumorigenic behavior, and which are therefore most likely to be responsive to inhibition with nilotinib. Based on these data, we suggest that a subset of appropriately-identified pediatric HGG should be considered candidates for nilotinib treatment.
References
Bondy ML, Scheurer ME, Malmer B, Barnholtz-Sloan JS, Davis FG, Il’yasova D, Kruchko C, McCarthy BJ, Rajaraman P, Schwartzbaum JA, Sadetzki S, Schloehofer B, Tihan T, Wiemels JL, Wrensch M, Buffler PA (2008) Brain tumor epidemiology: consensus from the Brain Tumor Epidemiology Consortium. Cancer 113(S7):1953–1968. doi:10.1002/cncr.23741
Pollack IF, Jakacki RI, Blaney SM, Hancock ML, Kieran MW, Phillips P, Kun LE, Friedman H, Packer R, Banerjee A, Geyer JR, Goldman S, Poussaint TY, Krasin MJ, Wang Y, Hayes M, Murgo A, Weiner S, Boyett JM (2007) Phase I trial of imatinib in children with newly diagnosed brainstem and recurrent malignant gliomas: a Pediatric Brain Tumor Consortium report. Neuro Oncol 9(2):145–160. doi:10.1215/15228517-2006-031
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (eds) (2007) WHO classification of tumours of the central nervous system, 4th edn. International Agency for Research on Cancer, Lyon
Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, Bax DA, Coyle B, Barrow J, Hargrave D, Lowe J, Gajjar A, Zhao W, Broniscer A, Ellison DM, Grundy RG, Baker SJ (2010) Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 28(18):3061–3068
Bax DA, Mackay A, Little SE, Carvalho D, Viana-Pereira Tamber N, Grigoriadis AE, Ashworth A, Reis RM, Ellison DM, Al-Saraj S, Hargrave D, Jones C (2010) A distinct spectrum of copy number aberrations in pediatric high-grade gliomas. Clin Cancer Res 16(12):3368–3377. doi:10.1158/1078-0432-CCR-10-0438
Qu H-Q, Jacob K, Fatet S, Ge B, Barnett D, Delattre O, Faury D, Montpetit A, Solomon L, Hauser P, Garami M, Bognar L, Hansely Z, Mio R, Farmer J-P, Albrecht S, Polychronakos C, Hawkins C, Jabado N (2010) Genome-wide profiling using single-nucleotide polymorphism arrays identifies novel chromosomal imbalances in pediatric glioblastomas. Neuro Oncol 12(2):153–163
Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley M-YC, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD (2010) Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res 70(2):512–519. doi:10.1158/0008-5472.can-09-1851
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jager N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Fruhwald MC, Roggendorf W, Kramm C, Durken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482(7384):226–231. doi:10.1038/nature10833nature10833
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Baker SJ (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44(3):251–253. doi:10.1038/ng.1102ng.1102
Khuong-Quang DA, Buczkowicz P, Rakopoulos P, Liu XY, Fontebasso AM, Bouffet E, Bartels U, Albrecht S, Schwartzentruber J, Letourneau L, Bourgey M, Bourque G, Montpetit A, Bourret G, Lepage P, Fleming A, Lichter P, Kool M, von Deimling A, Sturm D, Korshunov A, Faury D, Jones DT, Majewski J, Pfister SM, Jabado N, Hawkins C (2012) K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol 124(3):439–447. doi:10.1007/s00401-012-0998-0
Nakamura M, Shimada K, Ishida E, Higuchi T, Nakase H, Sakaki T, Konishi N (2007) Molecular pathogenesis of pediatric astrocytic tumors. Neuro Oncol 9(2):113–123. doi:10.1215/15228517-2006-036
Thorarinsdottir HK, Santi M, McCarter R, Rushing EJ, Cornelison R, Jales A, MacDonald TJ (2008) Protein expression of platelet-derived growth factor receptor correlates with malignant histology and PTEN with survival in childhood gliomas. Clin Cancer Res 14(11):3386–3394. doi:10.1158/1078-0432.ccr-07-1616
Faury D, Nantel A, Dunn SE, Guiot M-C, Haque T, Hauser P, Garami M, Bognar L, Hanzely Z, Liberski PP, Lopez-Aguilar E, Valera ET, Tone LG, Carret A-S, Del Maestro RF, Gleave M, Montes J-L, Pietsch T, Albrecht S, Jabado N (2007) Molecular profiling identifies prognostic subgroups of pediatric glioblastoma and shows increased YB-1 expression in tumors. J Clin Oncol 25(10):1196–1208. doi:10.1200/JCO.2006.07.8626
Haque T, Faury D, Albrecht S, Lopez-Aguilar E, Hauser P, Garami M, Hanzely Z, Bognar L, Del Maestro RF, Atkinson J, Nantel A, Jabado N (2007) Gene expression profiling from formalin-fixed paraffin-embedded tumors of pediatric glioblastoma. Clin Cancer Res 13(21):6284–6292. doi:10.1158/1078-0432.CCR-07-0525
Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E, Kung AL, Mestan J, Daley GQ, Callahan L, Catley L, Cavazza C, Azam M, Neuberg D, Wright RD, Gilliland DG, Griffin JD (2005) Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 7(2):129–141. doi:10.1016/j.ccr.2005.01.007
Manley PW, Stiefl N, Cowan-Jacob SW, Kaufman S, Mestan J, Wartmann M, Wiesmann M, Woodman R, Gallagher N (2010) Structural resemblances and comparisons of the relative pharmacological properties of imatinib and nilotinib. Bioorg Med Chem 18(19):6977–6986. doi:10.1016/j.bmc.2010.08.026
Verstovsek S, Giles FJ, Quintas-Cardama A, Manshouri T, Huynh L, Manley P, Cortes J, Tefferi A, Kantarjian H (2006) Activity of AMN107, a novel aminopyrimidine tyrosine kinase inhibitor, against human FIP1L1-PDGFR-alpha-expressing cells. Leuk Res 30(12):1499–1505. doi:10.1016/j.leukres.2006.03.012
Stover EH, Chen J, Lee BH, Cools J, McDowell E, Adelsperger J, Cullen D, Coburn A, Moore SA, Okabe R, Fabbro D, Manley PW, Griffin JD, Gilliland DG (2005) The small molecule tyrosine kinase inhibitor AMN107 inhibits TEL-PDGFRbeta and FIP1L1-PDGFRalpha in vitro and in vivo. Blood 106(9):3206–3213. doi:10.1182/blood-2005-05-1932
Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G (2007) Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 110(12):4055–4063. doi:10.1182/blood-2007-07-102061
White DL, Saunders VA, Dang P, Engler J, Zannettino AC, Cambareri AC, Quinn SR, Manley PW, Hughes TP (2006) OCT-1-mediated influx is a key determinant of the intracellular uptake of imatinib but not nilotinib (AMN107): reduced OCT-1 activity is the cause of low in vitro sensitivity to imatinib. Blood 108(2):697–704. doi:10.1182/blood-2005-11-4687
Shukla S, Skoumbourdis AP, Walsh MJ, Hartz AM, Fung KL, Wu CP, Gottesman MM, Bauer B, Thomas CJ, Ambudkar SV (2011) Synthesis and characterization of a BODIPY conjugate of the BCR-ABL kinase inhibitor Tasigna (nilotinib): evidence for transport of Tasigna and its fluorescent derivative by ABC drug transporters. Mol Pharm 8(4):1292–1302. doi:10.1021/mp2001022
Tiwari AK, Sodani K, Dai CL, Abuznait AH, Singh S, Xiao ZJ, Patel A, Talele TT, Fu L, Kaddoumi A, Gallo JM, Chen ZS (2013) Nilotinib potentiates anticancer drug sensitivity in murine ABCB1-, ABCG2-, and ABCC10-multidrug resistance xenograft models. Cancer Lett 328(2):307–317. doi:10.1016/j.canlet.2012.10.001
Hebron ML, Lonskaya I, Moussa CE (2013) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of alpha-synuclein in Parkinson’s disease models. Hum Mol Genet 22(16):3315–3328. doi:10.1093/hmg/ddt192ddt192
Verstovsek S, Akin C, Manshouri T, Quint´as-Cardama A, Huynha L, Manley P, Tefferi A, Cortes J, Giles FJ, Kantarjian H (2006) Effects of AMN107, a novel aminopyrimidine tyrosine kinase inhibitor, on human mast cells bearing wild-type or mutated codon 816 c-kit. Leuk Res 30:1365–1370. doi:10.1016/j.leukres.2006.04.005
Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, Ossenkoppele GJ, Nicolini FE, O’Brien SG, Litzow M, Bhatia R, Cervantes F, Haque A, Shou Y, Resta DJ, Weitzman A, Hochhaus A, le Coutre P (2007) Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 110(10):3540–3546. doi:10.1182/blood-2007-03-080689
Houghton PJ, Cheshire PJ, Hallman JD 2nd, Lutz L, Friedman HS, Danks MK, Houghton JA (1995) Efficacy of topoisomerase I inhibitors, topotecan and irinotecan, administered at low dose levels in protracted schedules to mice bearing xenografts of human tumors. Cancer Chemother Pharmacol 36(5):393–403
Trent J, Meltzer P, Rosenblum M, Harsh G, Kinzler K, Mashal R, Feinberg A, Vogelstein B (1986) Evidence for rearrangement, amplification, and expression of c-myc in a human glioblastoma. Proc Natl Acad Sci U S A 83(2):470–473
Bradley TR, Metcalf D (1966) The growth of mouse bone marrow cells in vitro. Aust J Exp Biol Med Sci 44(3):287–299
Liang M-L, Ma J, Ho M, Solomon L, Bouffet E, Rutka JT, Hawkins C (2008) Tyrosine kinase expression in pediatric high grade astrocytoma. J Neurooncol 87:247–253. doi:10.1007/s11060-007-9513-1
Paugh BS, Broniscer A, Qu C, Miller CP, Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS, Onar-Thomas A, Baker JN, Gajjar A, Ellison DW, Baker SJ (2011) Genome-wide analyses identify recurrent amplifications of receptor tyrosine kinases and cell-cycle regulatory genes in diffuse intrinsic pontine glioma. J Clin Oncol 29(30):3999–4006. doi:10.1200/jco.2011.35.5677
Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C (2010) Whole genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol 28(8):1337–1344. doi:10.1200/JCO.2009.25.5463
Frederick L, Wang X-Y, Eley G, James CD (2000) Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res 60(5):1383–1387
Liebermann T, Nusbaum H, Razon N, Kris R, Lax I, Soreg H, Whittle N, Waterfield M, Ullrich A, Schlessinger J (1985) Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 313(5998):144–147
Nilsson J, Thyberg J, Heldin CH, Westermark B, Wasteson A (1983) Surface binding and internalization of platelet-derived growth factor in human fibroblasts. Proc Natl Acad Sci U S A 80(18):5592–5596
Hart CE, Forstrom JW, Kelly JD, Seifert RA, Smith RA, Ross R, Murray MJ, Bowen-Pope DF (1988) Two classes of PDGF receptor recognize different isoforms of PDGF. Science 240(4858):1529–1531
Kashishian A, Kazlauskas A, Cooper JA (1992) Phosphorylation sites in the PDGF receptor with different specificities for binding GAP and PI3 kinase in vivo. EMBO J 11(4):1373–1382
Kazlauskas A, Kashishian A, Cooper JA, Valius M (1992) GTPase-activating protein and phosphatidylinositol 3-kinase bind to distinct regions of the platelet-derived growth factor receptor beta subunit. Mol Cell Biol 12(6):2534–2544
Berk BC, Alexander RW, Brock TA, Gimbrone MA Jr, Webb RC (1986) Vasoconstriction: a new activity for platelet-derived growth factor. Science 232(4746):87–90
Robson MC, Phillips LG, Thomason A, Robson LE, Pierce GF (1992) Platelet-derived growth factor BB for the treatment of chronic pressure ulcers. Lancet 339(8784):23–25. doi:10.1016/0140-6736(92)90143-Q
Schatteman GC, Morrison-Graham K, van Koppen A, Weston JA, Bowen-Pope DF (1992) Regulation and role of PDGF receptor alpha-subunit expression during embryogenesis. Development 115(1):123–131
Bax DA, Little SE, Gaspar N, Perryman L, Marshall L, Viana-Pereira M, Jones TA, Williams RD, Grigoriadis A, Vassal G, Workman P, Sheer D, Reis RM, Pearson AD, Hargrave D, Jones C (2009) Molecular and phenotypic characterisation of paediatric glioma cell lines as models for preclinical drug development. PLoS One 4(4):e5209. doi:10.1371/journal.pone.0005209
Takayama N, Sato N, O’Brien SG, Ikeda Y, Okamoto S (2002) Imatinib mesylate has limited activity against the central nervous system involvement of Philadelphia chromosome-positive acute lymphoblastic leukaemia due to poor penetration into cerebrospinal fluid. Br J Haematol 119(1):106–108
Acknowledgments
This work was supported by funding from brain tumour research assistance and information network (b.r.a.i.n.child) to GZ.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Au, K., Singh, S.K., Burrell, K. et al. A preclinical study demonstrating the efficacy of nilotinib in inhibiting the growth of pediatric high-grade glioma. J Neurooncol 122, 471–480 (2015). https://doi.org/10.1007/s11060-015-1744-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1744-y