
Abstract
The overexpression or amplification of the human epidermal growth factor receptor 2 gene (HER2/neu) is associated with high risk of brain metastasis (BM). The identification of patients at highest immediate risk of BM could optimize screening and facilitate interventional trials. We performed gene expression analysis using complementary deoxyribonucleic acid-mediated annealing, selection, extension and ligation and real-time quantitative reverse transcription PCR (qRT-PCR) in primary tumor samples from two independent cohorts of advanced HER2 positive breast cancer patients. Additionally, we analyzed predictive relevance of clinicopathological factors in this series. Study group included discovery Cohort A (84 patients) and validation Cohort B (75 patients). The only independent variables associated with the development of early BM in both cohorts were the visceral location of first distant relapse [Cohort A: hazard ratio (HR) 7.4, 95 % CI 2.4–22.3; p < 0.001; Cohort B: HR 6.1, 95 % CI 1.5–25.6; p = 0.01] and the lack of trastuzumab administration in the metastatic setting (Cohort A: HR 5.0, 95 % CI 1.4–10.0; p = 0.009; Cohort B: HR 10.0, 95 % CI 2.0–100.0; p = 0.008). A profile including 13 genes was associated with early (≤36 months) symptomatic BM in the discovery cohort. This was refined by qRT-PCR to a 3-gene classifier (RAD51, HDGF, TPR) highly predictive of early BM (HR 5.3, 95 % CI 1.6–16.7; p = 0.005; multivariate analysis). However, predictive value of the classifier was not confirmed in the independent validation Cohort B. The presence of visceral metastases and the lack of trastuzumab administration in the metastatic setting apparently increase the likelihood of early BM in advanced HER2-positive breast cancer.
Similar content being viewed by others

Introduction
The overexpression or amplification of the human epidermal growth factor receptor 2 gene (HER2/neu) is associated with high risk of brain metastasis (BM). Approximately 30–50 % of advanced HER2-positive breast cancer patients will develop BM, with an annual risk of around 10 % [1–5]. It has been speculated that improvements in systemic therapy resulting in greater numbers and more durable systemic responses may permit more time for BM relapse. Trastuzumab, a monoclonal antibody that targets the extracellular domain of HER2, is used in combination with chemotherapy to improve the survival of patients with HER2-positive tumors [6–10]. However, owing to its high molecular weight, penetration of trastuzumab into the central nervous system is extremely low, 1/420th of serum levels [11], and this compound is ineffective in treating established BM.
The development of BM predictors in advanced breast cancer patients might have practical clinical implications. First, the use of imaging to detect occult BM in unselected patients is controversial, whereas this strategy may be reasonable in patients at highest immediate risk. Second, reliable predictive factors may improve selection of patients in clinical trials assessing the efficacy of putative BM prevention strategies, such as prophylactic cranial irradiation or the use of brain-permeable compounds. Finally, these studies may prompt new therapeutic strategies.
In the present study we analyzed the risk of early BM according to gene expression, and clinical and pathological variables in two well annotated cohorts of advanced HER2-positive breast cancer patients.
Materials and methods
Patients
This study was approved by the Institutional Review Board of the coordinating centers (Medical University of Gdańsk, Poland and Indiana University, USA). Two patient cohorts were derived from a consecutive series of 315 advanced HER2-positive breast cancer patients treated in nine oncology centers in Poland and Serbia between 1993 and 2010 (consort diagram; Fig. 1). Discovery Cohort A (n = 167) and an independent validation Cohort B (n = 148) were collected between 2006–2008 and 2008–2010, respectively. According to standard clinical practice, no screening for occult BMs was used, therefore almost all BM were symptomatic. BM were defined as metastatic lesions involving the brain parenchyma, with or without accompanying leptomeningeal disease. Demographic and clinicopathologic data, as well as treatments and clinical follow-up were extracted from institutional databases or original patient files. Treatments were rule based (Table 1). Dominant metastatic sites were assigned into three categories: soft tissue, bones and viscera. Dominant metastatic site was classified by the category associated with the worst prognosis in the following order of increasing gravidity: soft tissue, bones, viscera [12].

Consort diagram
Pathology review
The starting material from each patient was a formalin-fixed, paraffin embedded specimen of primary breast cancer. A pre-cut section of each tumor, stained with hematoxylin and eosin, was reviewed by two pathologists (SB and WB) to confirm the presence of sufficient invasive breast cancer component (1 cm2 invasive tissue, ≥30 % tumor cells). In Cohorts A and B, 90/167 and 75/148 tumors, respectively, had sufficient material for molecular analysis. Expression of ER and PR was determined using immunohistochemistry (IHC), with 10 % of nuclear staining considered as a positive result. HER2 protein expression was determined using semiquantitative IHC (HercepTest, Dako A/S, Glostrup, Denmark) or HER-2/neuTest 4B5 (Ventana Medical Systems, Inc.). Only samples showing strong expression (scored 3+), defined as uniform, and intense membrane staining of at least 10 % of invasive tumor cells, were considered positive. The samples showing intermediate expression (scored 2+) were subjected to additional analysis of HER2 gene copy number using fluorescence in situ hybridization (FISH). Gene amplification by FISH was defined as a FISH ratio (HER2/centromeric probe for chromosome 17 ratio) of greater than 2.0. FISH-positive patients were considered HER2-positive.
RNA extraction
Tumor cells were processed using macrodissection to enrich their population for analysis. Sections were deparaffinized with CitriSolv clearing agent (Fisher Scientific Company, Fair Lawn, NJ) and scraped off from the slide into a microcentrifuge tube. Total RNA was extracted from three 10 μm thick whole tissue sections from each sample using the Roche high pure RNA paraffin kit according to manufacturer’s instructions (Roche Applied Science, Indianapolis, IN). Purified total RNA samples were stored frozen at −80 °C until needed for quality control (QC) analysis and subsequent gene expression profiling and quantitative reverse transcription PCR (qRT-PCR). The concentration of RNA was measured using Nanodrop® ND-1000 spectrophotometer (ThermoScientific, Wilmington, DE). RNA (200 ng) was reverse-transcribed to complementary deoxyribonucleic acid (cDNA) using iScript cDNA synthesis kit (Bio-Rad Laboratories, Inc., Hercules, CA). To prequalify RNA samples, SYBR Green-based qRT-PCR (Applied Biosystems, Foster City, CA) was performed for RPL13A ribosomal protein gene according to Illumina’s instructions (San Diego, CA).
DASL analysis
Cohort A samples were analyzed by annealing, selection, extension and ligation (DASL) assay using Cancer Panel v1 to provide expression data on 502 known cancer genes. DASL was performed with the Sentrix universal array (Illumina, San Diego, California) as per the manufacturer’s instructions [13] and blinded to patient outcome. Shortly, a 20-μl RT reaction containing a reaction mix (MMC; Illumina, San Diego, CA), biotinylated random hexamers and oligo-d(T)18, and total RNA, was incubated at room temperature for 10 min and then at 42 °C for 1 h. Pooled assay oligos were annealed to their sequence-specific targets on the cDNA under a controlled hybridization program. The cDNA was immobilized on paramagnetic beads and washed to remove any excess or mis-hybridized oligos. Hybridized oligos were then extended and ligated to generate amplifiable templates, using Illumina-supplied reagents and conditions (BeadStation User’s Manual, Illumina). A PCR reaction was performed with Cy3 labeled universal PCR primers. Single-stranded PCR products were prepared by denaturation, and were then hybridized to Sentrix arrays under a temperature gradient program. The arrays were imaged using a BeadArray Reader scanner (Illumina). The DASL assay was performed three times independently, and samples were hybridized to three different array matrices. The 502-gene assay was available in a 96-well format; this enabled analysis of all the samples in a single batch. Built-in internal controls and replicate samples were used to analyze stability of the assay. The r 2 values for the duplicate samples were greater than 0.95.
Generation of the 13-gene signature
Cohort A samples were divided into an internal training set and an internal testing set. Predictive analysis of microarray analysis (http://www-stat.stanford.edu/~tibs/PAM/) was performed to identify multigene profiles predictive for BM. The best gene-expression signature was selected based on a built-in 10-fold cross-validation analysis in PAM. Then the gene-signature was output as a single variable from the PAM. Its association with the BM free survival (BMFS) was analyzed in the internal testing set with a Cox regression analysis, in which clinical and demographic variable effects were justified. This analysis was performed with the R function, coxph. The gene signature construction from the internal training set used the optimal variable selection strategy in PAM, and p value was not considered. Then, the correlation between the gene signature and BMFS was assessed by the Cox regression model, and the p value <0.05 was considered as statistically significant.
Real-time qRT-PCR analysis
Owing to the abandoning of the 502-gene DASL assay by the manufacturer, and to increase the potential utility of the profile, we switched to a qRT-PCR assay. Apart from its clinical applicability, this method allows precise quantification of transcriptional abundance of identified genes. TaqMan reactions were performed in triplicates using custom array microfluidic cards preloaded with TaqMan gene expression assays containing 16 genes (13 discriminant genes and 3 reference genes) on an ABI Prism 7900HT fast real-time platform according to the manufacturer’s instructions. The primer sequences are listed in Table 2. Transferrin receptor (TFRC), beta cytoskeletal actin (ACTB) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were used as endogenous reference controls for normalization. Delta threshold cycle (ΔCt) values for each of the 13 genes of interest were normalized using the three endogenous reference controls according to the method of Applied Biosystem’s DataAssist™ Software. All procedures were performed blinded to patient outcomes. After normalization, \(2^{{ - \Updelta {\text{C}}_{\text{t}} }}\) values were subject to the leave-one-out cross-validated linear discriminant analysis (LDA), and coefficients for the individual genes were chosen. The coefficients for the individual genes and individual gene expression data for each patient were collated to develop an individual score, which was used for statistical analysis in both cohorts.
Statistical analysis
All statistical analyses were performed using STATA software version 11. Statistical significance was defined as p < 0.05. Gene expression data were normalized at the median level. Hierarchical clustering and singular value decomposition methods were applied to detect the outliers for QC purposes. The false discovery rate (FDR), an estimate of the proportion of errors committed by falsely rejecting null hypotheses was calculated for each gene. Categorical variables in both cohorts (including correlation of the developed 3-gene classifier with clinicopathologic variables) were compared using Pearson’s Chi squared test (χ 2). Survival curves were plotted using Kaplan–Meier method starting from date of primary breast cancer diagnosis to date of death or last follow-up. The BMFS was a primary endpoint and was defined from date of primary breast cancer diagnosis to date of BM diagnosis, death of any cause, or date of last follow-up. Univariate survival analysis and time to diagnosis of BM within 36 months in Cohorts A and B were performed with log-rank test, Wilcoxon test and Cox proportional hazard regression and controlled for the competing risk of death [14]. Multivariate analysis used a stepwise forward selection of univariate model with p ≤ 0.20.
Results
Characteristics and outcomes of study cohorts
Discovery Cohort A
Of the 84 primary tumors subjected to analysis in the Cohort A, 83 were analyzable (Fig. 1). The patient mean age was 48 years (range 30–64), with the patients roughly divided between pre- and post-menopausal status (Table 1). Eighty three percent of the tumors were invasive ductal cancers, 63 % were ER-negative and 71 % PR-negative. Ninety two percent of patients had dominant visceral metastatic disease; 98 % of patients received chemotherapy and 48 % endocrine therapy in adjuvant and/or metastatic setting. More than 40 % of patients received induction chemotherapy and 87 % of patients were administered trastuzumab in adjuvant or metastatic setting, usually in combination with chemotherapy. In 14 % of patients lapatinib was administered at trastuzumab relapse. Follow-up from breast cancer diagnosis varied from 1 to 185 months. Within this period, 48 patients developed symptomatic BM. The median time from initial breast cancer to BM diagnosis was 36 months (range 2–141 months). In 7 % of patients brain was the first site of distant relapse, with or without accompanying extracranial relapse. After BM, HER2 directed treatments included trastuzumab (33 % of patients), lapatinib (15 %) and either used sequentially (4 %). Seventy seven percent of patients received cranial radiotherapy. The median overall survival (OS) from the initial diagnosis of breast cancer was 44 months (range 0.9–185 months).
Validation Cohort B
The Cohort B, including 75 analyzable cases, was similar to Cohort A in terms of age, ER and PR expression, and tumor grade (Table 1). However, Cohort B included significantly more non-ductal cancers, patients were older at BM development and had different patterns of relapse (fewer distant relapses as the first failure, more first relapses in the visceral organs and fewer visceral dominant metastatic sites). Furthermore, more patients in this cohort received lapatinib following failure of trastuzumab. The median time to diagnosis of BM in this cohort was longer (40 months; range 0.33–125 months, compared to 36 months in Cohort A), and so was the median OS (50 months; range 11–186 months, compared to 44 months in Cohort A). In Cohort B 41 patients developed BM, including 16 that occurred within 36 months from diagnosis.
Determinants of BMFS and OS
Performed in Cohort A binary comparison for presence or absence of BM among 502 analyzed genes did not show any differential gene expression (25 having p < 0.05, a FDR of 1.0). However, a gene expression analysis in 22 and 21 patients who developed BM within 36 months (the median time to diagnosis of BM) versus thereafter, respectively, identified differential expression of 48 genes with p < 0.01 and FDR = 0.1. Predictive analysis of microarray analysis identified a 13-gene profile [cyclin dependent kinase 4 (CDK4), cyclin C (CCNC), focal adhesion kinase (protein tyrosine kinase 2, PTK2), v-myc avian myelocytomatosis viral oncogene homolog (MYC), BRCA1 associated RING domain 1 (BARD1), RAD51 homolog (RAD51), Fanconi anemia group G (FANCG), proliferating cell nuclear antigen (PCNA), papillary renal cell carcinoma-translocation associated (PRCC), translocated promoter region (TPR), cortactin (CTTN), desmoplakin (DSP), hepatoma-derived growth factor (HDGF)] at effectively distinguished patients with early versus late BM [hazard ratio (HR) 5.6, 95 % CI 1.9–16.5; p = 0.002 in the univariate analysis; HR 8.5, 95 % CI 2.6–28.0; p < 0.001 in the multivariate analysis; Table 3]. The microarray data have been deposited in NCBI’s gene expression omnibus (http://www.ncbi.nlm.nih.gov/geo; GSE38057).
In order to increase the potential clinical applicability of this signature, a qRT-PCR based analysis of the 13 genes (and 3 references) was performed and showed promising preliminary results [15, 16]. The TaqMan gene expression assay IDs for each gene was chosen to meet FFPE sample requirements for custom TLDA based on Applied Biosystems guidelines. As expected, DASL and qRT-PCR had inherent differences related to the platform (Fig. 2). As the next step, a leave-one-out LDA was performed using an updated database that had a longer follow-up (5 years) data. A predictive model that included only 3 of the original 13 genes: HDGF, RAD51 and TPR, with corresponding LDA coefficients of 1.06, 0.35 and −1.08, respectively, was developed. The 3-gene classifier was highly predictive of early BM both in univariate (HR 3.7, 95 % CI 1.3–11.1; p = 0.01) and multivariate analysis (HR 5.3, 95 % CI 1.6–16.7; p = 0.005; Table 3). High 3-gene classifier was associated with tumor grade 3, ER-negativity and less frequent use of endocrine treatment and trastuzumab in the adjuvant and/or metastatic setting (Table 4). Additionally, patients with high 3-gene classifier were more likely to develop the first relapse in the visceral organs.

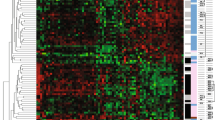
Cohort A. Distribution of the 13 gene transcript expression obtained from the RNA isolation process in DASL and qRT-PCR. a DASL (data was normalized using quantile normalization), b qRT-PCR normalized using the endogenous reference controls (ACTB, GAPDH, TFRC). The graph shows apparent inter-panel discordance of BARD1, CCNC and HDGF expression, and minor inter-panel discordance of FANCG and PCNA expression
In an independent Cohort B the mean qRT-PCR expression of 13 genes was different compared to Cohort A, and only 16 % of patients (compared to 59 % in Cohort A) were assigned to the high-risk group (Table 4). Accordingly, the 3-gene classifier was not predictive of early BM (HR 1.2, 95 % CI 0.3–20.0; p = 0.8; Table 3). In this cohort the high 3-gene classifier was associated with less frequent use of induction chemotherapy and more lung and liver metastases (Table 4).
In the multivariate analysis, in both cohorts the visceral location of first distant relapse (Cohort A: HR 7.4, 95 % CI 2.4–22.3; p < 0.001; Cohort B: HR 6.1, 95 % CI 1.5–25.6; p = 0.01) and the lack of trastuzumab administration in the metastatic setting (HR 5.0, 95 % CI 1.4–10.0; p = 0.009 and HR 10.0, 95 % CI 2.0–100.0; p = 0.008, respectively) correlated with early BM (Table 3). ER-negativity had a strong trend in Cohort A (HR 2.8, 95 % CI 0.9–9.1; p = 0.07) and was significant in Cohort B (HR 5.0, 95 % CI 1.1–10.0; p = 0.04).
In both cohorts the independent variables associated with shorter OS included higher tumor grade (HR 1.9, 95 % CI 1.1–3.3; p = 0.02; HR 1.9, 95 % CI 1.4–3.2; p = 0.03), ER negativity (HR 2.0, 95 % CI 1.1–3.3; p = 0.03; HR 2.5, 95 % CI 1.4–5.0; p < 0.01) and the lack of primary tumor surgery (HR 5.0, 95 % CI 2.0–10.0; p < 0.01; HR 3.3, 95 % CI 1.7–10.0; p < 0.01).
Discussion
The aim of this study was to identify molecular predictors of the BM development in advanced HER2-positive breast cancer patients. This subset of breast cancer patients carry particularly high risk of BM. Additionally, some studies suggested increased risk of BM associated with the use of trastuzumab [17].
The current study employed a high throughput DASL technology based on the expression of 502 cancer related genes in addition to analysis of the clinicopathologic variables. This targeted gene analysis did not demonstrate any differential gene expression in patients who did and did not develop BM. This may likely be due to the limited number of genes analyzed, but it is also possible that BM in advanced HER2-positive breast cancer patients is a biologically determined, stochastic and inevitable event. Further analysis of the DASL led to identification of a 13-gene profile that was apparently predictive for development of early BM [15]. For precise quantification of transcriptional abundance of identified genes, we employed qRT-PCR technology, which identified a 3-gene classifier (RAD51, HDGF, TPR), also seemingly predictive for early BM. However, the significance of this classifier was not confirmed in the independent cohort.
The retrospective design of this study made it difficult to control for major clinicopathologic differences between Cohorts A and B. In consequence, patients in Cohort B had fewer ductal carcinomas and, even more importantly, less frequently received neoadjuvant chemotherapy. Gene expression alterations of breast cancer were recently demonstrated to be drug-specific, and drug-induced tumor gene signatures may be more informative than unchallenged signatures in predicting treatment outcomes [18, 19]. The study by Bos et al. [20] showed that BM gene set tested in various breast cancer cohorts was less BM predictive in patients whom received postoperative systemic therapy compared to those whom did not. This confirms the hypothesis that systemic therapies, apart from their preventive effect, may also alter the pattern of relapse in breast cancer. In this study, patients in Cohort B, compared to Cohort A, had also infrequent first relapse at distant sites and significantly fewer visceral metastases. Furthermore, much more patients in this cohort received lapatinib at trastuzumab relapse (32 %, compared to 14 % in Cohort A). The pivotal study by Geyer et al. [21] showed that the addition of lapatinib to capecitabine after progression on trastuzumab resulted in decreased BM occurrence, and preclinical studies show that lapatinib prevents BMs formation by 53 % in a HER2-transfected model system [22]. The abovementioned differences between both cohorts led to better general prognosis in Cohort B compared to Cohort A, expressed by longer OS and time to diagnosis of BM. Finally, the imbalanced proportion of patients with high gene classifier in both cohorts (59 % in Cohort A vs. 16 % in Cohort B) might have largely impacted study results.
Although the gene signature could not be validated, it identified a number of genes that could be important in the development of BM. The most important of which is RAD51, a gene involved in homologous recombination in DNA double strand breaks repair [20]. RAD51 expression has been linked to response to neoadjuvant therapy [23–25]. We have previously reported that high cytoplasmic expression of RAD51 in breast cancer is associated with significantly increased risk of BM, particularly in combination with high Ki-67 index and ER-negativity [26]. Further, in other study demonstrated that BARD1 and RAD51 are frequently overexpressed in BMs from breast cancer and may constitute a mechanism to overcome reactive oxygen species-mediated genotoxic stress in the metastatic brain [27]. Taken together, this data suggest that RAD51 targeting might be important in HER2-positive breast cancer. High nuclear expression of HDGF, another gene constituting our 3-gene signature, was earlier found to associate with high tumor grade, Ki-67 >20 %, lymph node involvement and poor prognosis in breast cancer patients [28, 29]. Chen et al. [29] demonstrated that nuclear HDGF over-expression stimulates epithelial–mesenchymal transition of breast cancer cells by down-regulation of E-cadherin and up-regulation of vimentin. The third gene of our signature—TPR, a translocated promoter region nuclear basket protein, is poorly characterized but has a normal function in nuclear pore function and is the target of oncogenic fusions [30].
In the current study, the clinical factors associated with early development of BM were visceral location of first relapse and, at a borderline level, ER-negativity, the two hallmarks of tumor aggressiveness. This is partly consistent with our earlier study in advanced HER2-positive breast cancer patients, showing the association between the risk of BM and shorter time to first extracranial progression [5]. The association between ER-negativity and the occurrence of BM in HER2-positive breast cancer patients was earlier reported by other authors [2, 4, 31, 32]. Indeed, the clinical behavior including tumor kinetics and sites of recurrence in ER-positive/HER2 positive (HER2-positive luminal B) breast cancer is different compared to that in non-luminal HER2 enriched subtype [31–34]. We also showed that trastuzumab administration in the metastatic setting may reduce the risk of early BM. This is in line with two other studies, that noticed shorter time to development of BM in HER2-positive patients who never received trastuzumab [35, 36].
Conclusions
We demonstrated that the presence of visceral metastases and the lack of trastuzumab administration in the metastatic setting apparently increase the likelihood of early BM in advanced HER2-positive breast cancer, and the 3-gene classifier does not improve their predictive value. Our study also illustrates the difficulties in developing clinically useful predictive markers in the retrospective setting [37]. In our case these included problems associated with archival tissue collection, heterogeneity of patient populations and inconsistent therapeutic approaches over the study period. Further studies, including larger and more homogeneous groups, are necessary to identify biomarkers, which may help in designing BM preventive trials and prompt new treatment strategies.
References
Bendell J, Domchek S, Burstein H, Harris L, Younger J, Kuter I, Bunnell C, Rue M, Gelman R, Winer E (2003) Central nervous system metastases in women who receive trastuzumab-based therapy for metastatic breast carcinoma. Cancer 97:2972–2977
Clayton A, Danson S, Jolly S, Ryder WD, Burt PA, Stewart AL, Wilkinson PM, Welch RS, Magee B, Wilson G, Howell A, Wardley AM (2004) Incidence of cerebral metastases in patients treated with trastuzumab for metastatic breast cancer. Br J Cancer 91:639–643
Stemmler HJ, Kahlert S, Siekiera W, Untch M, Heinrich B, Heinemann V (2006) Characteristics of patients with brain metastases receiving trastuzumab for HER2 overexpressing metastatic breast cancer. Breast 15:219–225
Brufsky A, Mayer M, Rugo H, Kaufman PA, Tan-Chiu E, Tripathy D, Tudor IC, Wang LI, Brammer MG, Shing M, Yood MU, Yardley DA (2011) Central nervous system metastases in patients with HER2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin Cancer Res 17:4834–4843
Duchnowska R, Dziadziuszko R, Czartoryska-Arlukowicz B, Radecka B, Szostakiewicz B, Sosińska-Mielcarek K, Karpińska A, Starosławska E, Kubiatowski T, Szczylik C (2009) Risk factors for brain relapse in HER2-positive metastatic breast cancer patients. Breast Cancer Res Treat 117:297–303
Slamon D, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344:783–792
Romond EH, Perez EA, Bryant J, Suman VJ, Geyer CE Jr, Davidson NE, Tan-Chiu E, Martino S, Paik S, Kaufman PA, Swain SM, Pisansky TM, Fehrenbacher L, Kutteh LA, Vogel VG, Visscher DW, Yothers G, Jenkins RB, Brown AM, Dakhil SR, Mamounas EP, Lingle WL, Klein PM, Ingle JN, Wolmark N (2005) Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med 353:1673–1684
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, Gianni L, Baselga J, Bell R, Jackisch C, Cameron D, Dowsett M, Barrios CH, Steger G, Huang CS, Andersson M, Inbar M, Lichinitser M, Láng I, Nitz U, Iwata H, Thomssen C, Lohrisch C, Suter TM, Rüschoff J, Suto T, Greatorex V, Ward C, Straehle C, McFadden E et al (2005) Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 353:1659–1672
Joensuu H, Kellokumpu-Lehtinen PL, Bono P, Alanko T, Kataja V, Asola R, Utriainen T, Kokko R, Hemminki A, Tarkkanen M, Turpeenniemi-Hujanen T, Jyrkkiö S, Flander M, Helle L, Ingalsuo S, Johansson K, Jääskeläinen AS, Pajunen M, Rauhala M, Kaleva-Kerola J, Salminen T, Leinonen M, Elomaa I, Isola J, FinHer Study Investigators (2006) Adjuvant docetaxel or vinorelbine with or without trastuzumab for breast cancer. N Engl J Med 354:809–820
Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, Mackey J, Glaspy J, Chan A, Pawlicki M, Pinter T, Valero V, Liu MC, Sauter G, von Minckwitz G, Visco F, Bee V, Buyse M, Bendahmane B, Tabah-Fisch I, Lindsay MA, Riva A, Crown J, Breast Cancer International Research Group (2011) Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med 365:1273–1283
Stemmler H-J, Schmitt M, Willems A, Bernhard H, Harbeck N, Heinemann V (2007) Ratio of trastuzumab levels in serum and cerebrospinal fluid is altered in HER2-positive breast cancer patients with brain metastases and impairment of the blood–brain barrier. Anticancer Drugs 18:23–28
Rozencweig M, Heuson JC (1975) Breast cancer: prognostic factors and clinical evaluation. In: Staquet MJ (ed) Cancer Therapy: prognostic factors and criteria of response. Raven Press, New York, p 171
Bibikova M, Talantov D, Chudin E, Yeakley JM, Chen J, Doucet D, Wickham E, Atkins D, Barker D, Chee M, Wang Y, Fan JB (2004) Quantitative gene expression profiling in formalin-fixed, paraffin-embedded tissues using universal bead arrays. Am J Pathol 165:1799–1807
Scrucca L, Santucci A, Aversa F (2007) Competing risk analysis using R: an easy guide for clinicians. Bone Marrow Transplant 40:381–387
Duchnowska R, Jassem J, Thorat M, Morimiya A, Li L, Sledge GW Jr, Biernat W, Szczylik C, Steeg P, Badve S (2008) Gene expression analysis for prediction of rapid brain metastasis in HER2-positive breast cancer patients [abstract]. J Clin Oncol 26:s45
Duchnowska R, Jassem J, Pankaj Goswami C, Gokmen-Polar Y, Li L, Thorat MA, Flores N, Hua E, Woditschka S, Palmieri D, Steinberg SM, Biernat W, Sosinska-Mielcarek K, Szostakiewicz B, Czartoryska-Arlukowicz B, Radecka B, Tomasevic Z, Sledge GW, Steeg PS, Badve SS, Polish Brain Metastasis Consortium (2012) 13-Gene signature to predict rapid development of brain metastases in patients with HER2-positive advanced breast cancer [abstract]. J Clin Oncol 30(Suppl):505
Duchnowska R, Szczylik C (2005) Central nervous system metastases in breast cancer patients administered trastuzumab. Cancer Treat Rev 31:312–318
Lee SC, Xu X, Lim YW, Iau P, Sukri N, Lim SE, Yap HL, Yeo WL, Tan P, Tan SH, McLeod H, Goh BC (2009) Chemotherapy-induced tumor gene expression changes in human breast cancers. Pharmacogenet Genomics 19:181–192
Lee SC, Xu X, Chng WJ, Watson M, Lim YW, Wong CI, Iau P, Sukri N, Lim SE, Yap HL, Buhari SA, Tan P, Guo J, Chuah B, McLeod HL, Goh BC (2009) Post-treatment tumor gene expression signatures are more predictive of treatment outcomes than baseline signatures in breast cancer. Pharmacogenet Genomics 19:833–842
Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, van de Vijver MJ, Gerald WL, Foekens JA, Massagué J (2009) Genes that mediate breast cancer metastasis to the brain. Nature 459:1005–1009
Geyer CE, Forster J, Lindquist D, Chan S, Romieu CG, Pienkowski T, Jagiello-Gruszfeld A, Crown J, Chan A, Kaufman B, Skarlos D, Campone M, Davidson N, Berger M, Oliva C, Rubin SD, Stein S, Cameron D (2006) Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med 355:2733–2743
Gril B, Palmieri D, Bronder JL, Herring JM, Vega-Valle E, Feigenbaum L, Liewehr DJ, Steinberg SM, Merino MJ, Rubin SD, Steeg PS (2008) Effect of lapatinib on the outgrowth of metastatic breast cancer cells to the brain. J Natl Cancer Inst 100:1092–1103
Kim TM, Ko JH, Hu L, Kim SA, Bishop AJ, Vijg J, Montagna C, Hasty P (2012) RAD51 mutants cause replication defects and chromosomal instability. Mol Cell Biol 32:3663–3680
Graeser M, McCarthy A, Lord C, Savage K, Hills M, Salter J, Orr N, Parton M, Smith IE, Reis-Filho JS, Dowsett M, Ashworth A, Turner NC (2010) A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin Cancer Res 16:6159–6168
Asakawa H, Koizumi H, Koike A, Takahashi M, Wu W, Iwase H, Fukuda M, Ohta T (2010) Prediction of breast cancer sensitivity to neoadjuvant chemotherapy based on the status of DNA damage repair proteins. Breast Cancer Res 12:R17
Sosińska-Mielcarek K, Duchnowska R, Winczura P, Badzio A, Majewska H, Lakomy J, Pęksa R, Pieczyńska B, Radecka B, Dębska S, Biernat W, Jassem J (2013) Immunohistochemical prediction of brain metastases in patients with advanced breast cancer: the role of RAD51. Breast 22:1178–1183
Woditschka S, Evans L, Duchnowska R, Reed LT, Palmieri D, Qian Y, Badve S, Sledge G Jr, Gril B, Aladjem MI, Fu H, Flores NM, Gökmen-Polar Y, Biernat W, Szutowicz-Zielińska E, Mandat T, Trojanowski T, Och W, Czartoryska-Arlukowicz B, Jassem J, Mitchell JB, Steeg PS (2014) DNA double-strand break repair genes and oxidative damage in brain metastasis of breast cancer. J Natl Cancer Inst 106(7):dju145
Chen X, Yun J, Fei F, Yi J, Tian R, Li S, Gan X (2012) Prognostic value of nuclear hepatoma-derived growth factor (HDGF) localization in patients with breast cancer. Pathol Res Pract 208:437–443
Chen SC, Kung ML, Hu TH, Chen HY, Wu JC, Kuo HM, Tsai HE, Lin YW, Wen ZH, Liu JK, Yeh MH, Tai MH (2012) Hepatoma-derived growth factor regulates breast cancer cell invasion by modulating epithelial–mesenchymal transition. J Pathol 228:158–169
Bangs PL, Sparks CA, Odgren PR, Fey EG (1996) Product of the oncogene-activating gene Tpr is a phosphorylated protein of the nuclear pore complex. J Cell Biochem 61:48–60
Vaz-Luis I, Ottesen RA, Hughes ME, Marcom PK, Moy B, Rugo HS, Theriault RL, Wilson J, Niland JC, Weeks JC, Lin NU (2012) Impact of hormone receptor status on patterns of recurrence and clinical outcomes among patients with human epidermal growth factor-2-positive breast cancer in the National Comprehensive Cancer Network: a prospective cohort study. Breast Cancer Res 14:R129
Kennecke H, Yerushalmi R, Woods R, Cheang MC, Voduc D, Speers CH, Nielsen TO, Gelmon K (2010) Metastatic behavior of breast cancer subtypes. J Clin Oncol 28:3271–3277
Paluch-Shimon S, Ben-Baruch N, Wolf I, Zach L, Kopolovic J, Kruglikova A, Modiano T, Yosepovich A, Catane R, Kaufman B (2009) Hormone receptor expression is associated with a unique pattern of metastatic spread and increased survival among HER2-overexpressing breast cancer patients. Am J Clin Oncol 32:504–508
Vaz-Luis I, Winer EP, Lin NU (2013) Human epidermal growth factor receptor-2-positive breast cancer: does estrogen receptor status define two distinct subtypes? Ann Oncol 24:283–291
Dawood S, Broglio K, Esteva FJ, Ibrahim NK, Kau SW, Islam R, Aldape KD, Yu TK, Hortobagyi GN, Gonzalez-Angulo AM (2008) Defining prognosis for women with breast cancer and CNC metastases by HER2 status. Ann Oncol 19:1242–1248
Park IH, Ro J, Lee KS, Nam BH, Kwon Y, Shin KH (2009) Trastuzumab treatment beyond brain progression in HER2-positive metastatic breast cancer. Ann Oncol 20:56–62
Diamandis EP (2010) Cancer biomarkers: can we turn recent failures into success? J Natl Cancer Inst 102:1462–1467
Acknowledgments
This work was supported by the US Department of Defense Breast Cancer Research Program, Grant Number W81 XWH-062-0033. Additional support was from the Intramural Research Programs of the National Cancer Institute in Bethesda, USA and Medical University of Gdańsk, Poland.
Conflict of interest
GWS and SB were supported by the Susan G Komen For the Cure®. The authors indicated no potential competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Duchnowska, R., Jassem, J., Goswami, C.P. et al. Predicting early brain metastases based on clinicopathological factors and gene expression analysis in advanced HER2-positive breast cancer patients. J Neurooncol 122, 205–216 (2015). https://doi.org/10.1007/s11060-014-1704-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-014-1704-y