
Abstract
Insects in Tenebrionidae have unique stress adaptations that allow them to survive temperature extremes. We report here a gene expression profiling of Microdera punctipennis, a beetle in desert region, to gain a global view of its environmental adaptations. A total of 48,158,004 reads were obtained by transcriptome sequencing, and the de novo assembly yielded 56,348 unigenes with an average length of 666 bp. Based on similarity searches with a cut-off E-value of 10−5 against two protein sequence databases, 41,109 of the unigenes (about 72.96 %) were matched to known proteins. An in-depth analysis of the data revealed a large number of genes were associated with environmental stress, including genes that encode heat shock proteins, antifreeze proteins, and enzymes such as chitinase, trehalose, and trehalose-6-phosphate synthase. This study generated a substantial number of M. punctipennis transcript sequences that can be used to discover novel genes associated with stress adaptation. These sequences are a valuable resource for future studies of the desert beetle and other insects in Tenebrionidae. Transcriptome analysis based on Illumina paired-end sequencing is a powerful approach for gene discovery and molecular marker development for non-model species.
Similar content being viewed by others

Introduction
Deserts are among the most hostile habitats on earth. In summer, it is extremely hot during daytime, while in the depth of the winter’s night, it is surprisingly cold, as well as extremely dry in some seasons. Under these extreme conditions, small arthropods and particularly Tenebrionidae beetles are conspicuous components of the fauna. To achieve this impressive resistance to extreme stress, these small animals possess several behavioral, morphological and physiological adaptations [1–3], such as burying themselves deeply in the substrate to avoid high temperatures and extreme dry during the day [1, 4], and taking up fog-water as a water source [5–8]. Most desert tenebrionids adopt seasonal behavioral changes to avoid hostile conditions [9, 10]. Subelytral cavity, an airtight space formed by the fusion of the elytra [11, 12], is found especially in desert Tenebrionidae and it helps to lower cuticular water permeability in desert beetles [13, 14].
Desert insects have the capacity of making significant and rapid adjustments to even slight changes in environmental temperature in their physiological state, characterized by cellular desiccation, build-up of metabolic wastes and depressed metabolic activity [15]. However, even in the frozen state some complex physiological processes continue, including cryoprotectant synthesis [16, 17] and diapause development [18]. Understanding of the roles of various proteins in insect has advanced substantially in the past 20 years. The development of powerful molecular tools and the increasing ease of their application have facilitated the identification and structural characterization of novel proteins, and progress is being made on determining their function in promoting winter survival in insects. Heat shock proteins (HSPs), also known as stress proteins, play a critical role in protecting organisms from injury due to high or low temperature [19], anoxia, desiccation [20] and a range of chemical stresses [21]. Besides, it is well known that antifreeze proteins (AFPs) play important roles in protecting poikilothermic organisms from freezing by promoting supercooling and inhibiting ice formation [22]. Moreover, it is found that AFP genes also expressed in summer beetles in desert region [23, 24] and is induced by high temperature [25]. These results suggest that AFPs may play a role in the adaptation of desert insects to environment.
One of these, Microdera punctipennis (Coleoptera: Tenebrionidae) is an endemic beetle in the Gurbantunggut Desert in Xinjiang [26], the north west of China. It is flightless, night active; and its behavioral and morphological characteristics for desert living have been identified carefully [10]. The day-night and seasonal temperature vary greatly in this region. This extreme variation in temperature might suggest that M. punctipennis have evolved a range of physiological and molecular adaptations for survival. Adults of M. punctipennis have supercooling points below −19.6 °C, and their capacity for supercooling has been shown to increase considerably with decreasing of water in their body fluid, but the underlying molecular basis remains unknown [23]. The study of desert beetles is important because it illustrates many of the solutions evolved by arthropods to the problems engendered, in an extreme form, by life in all terrestrial environments.
RNA-Seq is a recently developed large-scale genome-wide approach that has been applied successfully to gene discovery and expression profiling, and to the study of functional, comparative and evolutionary genomics in non-model organisms for which little previous genomic information existed. RNA-Seq has the advantages of being cost effective, highly sensitive, and accurate, with a large dynamic range [27]. In the past few years, this technology has been used to investigate molecular mechanisms in insect species such as Micrarchus nov. sp. 2, Tomicus yunnanensis, and Cryptolaemus montrouzieri [28–30]. Here, we describe the use of Illumina/Solexa paired-end technology for de novo transcriptome analysis of M. punctipennis. We obtained transcriptome sequences and discovered most of the known HSP and AFP genes, as well as the genes involved in the pathways for trehalose and chitin biosynthesis. Here, for the first time, we report the genomic profile information of the arid beetle M. punctipennis. This study also provides an insight into the molecular pathways involved in stress adaptation in this species.
Experimental procedures
Insects
M. punctipennis beetles were collected from the southern edge of the Gurbantunggut Desert (N 44°24, E 087°51′, 444 m), Xinjiang, China. The M. punctipennis adults were reared at 25 °C in the laboratory. Then, the samples were frozen in liquid nitrogen and stored at −80 °C until further use.
cDNA library generation and Illumina sequencing
Total RNA was extracted from three adult beetles using TRIzol Reagent (Sangon Biotech, China) according to the manufacturer’s instructions. The extracted RNA was assessed for quality and quantified using an Agilent 2100 Bioanalyzer (Agilent Technologies, Mississauga, Canada) with an RNA integration number (RIN) of 8, which is an algorithm for assigning integrity values to RNA measurements. For transcriptome analysis, the cDNA library was prepared using the TruSeq Sample Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturer’s recommendations. Briefly, mRNA was purified from 2 μg of total RNA using oligo (dT) magnetic beads. Divalent cations were used to fragment the purified mRNA into small pieces at 94 °C for 5 min; thereby priming bias was avoided when synthesizing the cDNA. The cleaved RNA fragments were used for double-stranded cDNA synthesis using a SuperScript Double-Stranded cDNA Synthesis kit (Invitrogen, Camarillo, CA, USA) with random hexamer (N6) primers (Illumina). The synthesized cDNA was subjected to end repair and a-Tailing processes before ligation of the adaptors. The end products were purified using a 2 % TAE-agarose gel (Certified LowRange Ultra Agarose, Bio-Rad) and enriched by PCR to create the final cDNA library with sequences of approximately 300 bp. After detection using an Agilent 2100 Bioanalyzer, the cDNA library clusters were generated by cBot machine (Illumina, San Diego, CA, USA) and then sequenced in Pair-End method by Sangon Biotech (Shanghai) Co., Ltd., China using an Illumina HiSeqTM 2000 (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions.
Sequence statistic and de novo assembly
Prior to assembly, the raw reads were cleaned by removing adapter sequences through the standard Illumina pipeline including the CASSAVA program (http://support.illumina.com/sequencing/sequencing_software/casava.ilmn). Low quality reads (those with quality value less than 20) and reads containing N (N represents ambiguous bases in reads), length less than 35 bp were filtered by a sliding window approach, the window size is 5 bp [31]. De novo assembly of the valid reads was performed using the November 2011 version of the Trinity program (http://trinityrnaseq.sourceforge.net/) which was designed specifically for transcriptome assembly from RNA-Seq data [32]. Briefly, Trinity combines reads of a certain length of overlap to form longer fragments and then processes them for sequence clusters with the sequence clustering software TGICL. The resultant sequences were defined as unigenes.
Bioinformatic analysis
The assembled unigenes were searched against the NCBI nr sequence database (ftp://ncbi.nih.gov), the Swiss-Prot database (http://web.expasy.org/docs/swiss-prot_guideline.html), kyoto encyclopedia of genes and genome (KEGG, http://www.genome.jp/kegg/), cluster of orthologous groups (COG) and eukaryotic orthologous groups (KOG) (ftp://ncbi.nih.gov/pub/COG/COG) with the BLASTX algorithm (accessed in Sept 2012). The E-value cut-off was set at 10−5. Genes were tentatively identified based on the best hits against known sequences. Blast2GO [33] was used to predict the functions of the sequences, to assign gene ontology (GO) terms (http://www.geneontology.org/), and to predict the metabolic pathways in COG and KEGG databases. Amino acid sequences were deduced by using ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and GENSCAN (http://genes.mit.edu/GENSCAN.html). The putative protein sequences were used for alignment by ClustalX (v1.83) program [34]. The MEGA5.0 software [35] was used to construct the consensus phylogenetic tree by using the neighbor-joining method based on Poisson correction model. Bootstrap analysis of 1,000 replication trees was performed to evaluate the branch strength of each tree.
Results
Illumina high-throughput sequencing and de novo assembly
A total of 48,158,004 raw reads were obtained by HiSeq™ 2000 (Illumina) paired-end sequencing (Table 1). After a stringent filtering process 39,654,340 valid reads of average length 95 bp were obtained.
We used the Trinity software to perform a paired end-joining de novo assembly of the valid reads. After assembly, 56,348 unigenes with an average length of 666 bp and an N50 of 1,603 bp were obtained. Of the 56,348 unigenes, 11,568 unigenes (20.52 %) were >1,000 bp long and 2,014 (3.57 %) were >3,000 bp long.
Annotation and function assignment
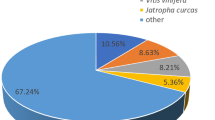
To identify putative functions, the 56,348 unigenes were firstly aligned by BLASTX (E-value ≤ 10−5) to several protein databases: NCBI nr, UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, CDD and Pfam. A total of 41,109 (72.96 %) unigenes had at least one hit to one of the databases (Table 2) and quite a large proportion (about 30 %) apparently has no significant match to any of the sequences in these databases, indicating that they may contain novel sequences and, perhaps, a high number of Coleoptera or species-specific transcripts or transcript parts (e.g. orphan UTRs). This might be expected, because there is very little sequence information from species closely related to M. punctipennis in these databases. The species distribution of the best match result for each sequence showed that the M. punctipennis sequences have 64.56 % matches with sequences from the Coleoptera species (Tribolium castaneum) (Fig. 1), while very low proportion (<1 %) of them have matches to other insects, for example, there was only 0.27 % (number of unigenes were 94) of them have matches to Drosophila melanogaster (not show independently in the figure). It demonstrated that M. punctipennis has a near evolution distance with T. castaneum.

Species distribution of the BLAST hit for each unigenes. Note that nearly 64.56 % of top hits are to the beetle T. castaneum whose complete genome has been sequenced. We used the first hit of each sequence for analysis
Pathway annotation was carried out based on the GO, COG/KOG (KOG is the eukaryotic version of COG), and KEGG databases.
Assignment of GO terms
GO (http://www.geneontology.org/) is an international classification system for standardized gene functions, offering a controlled vocabulary and a strictly defined conceptualization for comprehensively describing the properties of genes and their products within any organism [36]. The three main, independent GO categories are biological processes, molecular functions, and cellular components. A total of 8,477 different GO terms were assigned to 27,823 predicted unigene-encoded peptides that previously had matches with known proteins in the UniProtKB database. The terms were from the three main GO categories and covered 52 sub-categories (functional groups) (Fig. 2). Within the biological process group, the majority of unigenes were related to metabolic process (18678, 21.16 %) and cellular process (18195, 21.05 %); within cellular component, the largest proportion were assigned to cells (19422, 29.49 %), cell part (19422, 29.42 %), and organelles (10828, 16.44 %); and within molecular function the majority were assigned as binding (20084, 40.12 %) and catalytic activity (17818, 35.59 %) including hydrolases, kinases, and transferases, allowing for the identification of genes that may be involved in secondary metabolite synthesis pathways.

Pie charts showing gene ontology (GO) classification (level 2). GO analysis of Mp sequences corresponding to 27, 823 unigenes, as predicted for their involvement in biological processes (a) cellular component (b) and molecular function (c) is known
COG/KOG classification
COG (http://www.ncbi.nlm.nih.gov/COG/) compares the protein sequences that are encoded in complete genomes and represents them in major phylogenetic lineages [37]. The COG construction protocol included an automatic procedure for detecting candidate sets of orthologs, manual splitting of multidomain proteins into the component domains, and subsequent manual curation and annotation [38]. Furthermore, it has been extended to complex, multicellular eukaryotes by constructing clusters of probable orthologs [39]. Altogether, 8,980 unigenes was clustered into 25 functional categories (Fig. 3a). Among of them, the ‘‘general function prediction only’’ cluster was the largest (16.82 %), followed by ‘‘function unknown’’ (11.82 %). The other larger categories were: (posttranslational modification, protein turnover, chaperones (7.01 %); replication, recombination and repair (6.39 %) amino acid transport and metabolism (6.10 %); inorganic ion transport and metabolism (5.76 %); and cell cycle control, cell division, chromosome partitioning (4.91 %). An additional 649 unigenes (4.02 %) belonged to the “carbohydrate transport and metabolism” group among which 17 unigenes were annotated as chitinase. The COG classifications shed some light on specific responses and functions of genes that may be involved in regulating various molecular processes in M. punctipennis. The KOG classifications corresponded to 25 of the functional categories already observed in the COG analysis (Fig. 3b).

Histogram presenting clusters of orthologous group (COG/KOG) classification. a Of 56, 348 unigenes, 8, 980 sequences were assigned to 25 COG classification, b Of 56, 348 unigenes, 18, 014 sequences were assigned to 25 KOG classification
Assignment of KEGG pathways
To identify the biological pathways that are active in the M. punctipennis, we mapped the 56,348 annotated sequences to the reference canonical pathways in KEGG. A total of 9,986 unigenes were assigned to 283 known metabolic or signaling KEGG pathways. The top 10 KEGG pathways were spliceosome (290 unigenes), purine metabolism (269), protein processing in endoplasmic reticulum (261), Huntington’s disease (239), lysosome (227), RNA transport (225), ubiquitin mediated proteolysis (221), pathways in cancer (218), endocytosis (208), and focal adhesion (204). These annotations will provide a valuable resource for investigating specific processes, functions and pathways in M. punctipennis.
Several of the KEGG metabolite pathways were implicated in enhancing stress defense through their generation of specific metabolites. Among the 9,986 unigenes, 1,689 were mapped to 35 pathways that are related to metabolism (Fig. 4). For example, the “purine metabolism” (ID: ko00230) and “amino sugar and nucleotide sugar metabolism” (ID: ko00520) pathways were the largest groups, containing a total of 425 unigenes among them. A further 142 and 113 unigenes were assigned to the “glycerophospholipid metabolism” (ID: ko00564) and “aminoacyl-tRNA biosynthesis” (ID: ko00970) pathways, respectively.

Unigenes from M. punctipennis related to metabolic pathways
Putative environment stress-related unigenes
Heat shock proteins
A total of 72 HSP-related unigenes were identified in the M. punctipennis transcriptome and 31 of them were longer than 500 bp (Table 3). The majority of the HSP-related unigenes were predicted to encode the HSP70 type. The other HSP types among the HSP-related unigenes were, HSP1, HSP9, HSP20.6, HSP90, HSP60, sHSP21, and HSP cognate 1. These results should be validated by gene cloning based on the fragments obtained here.
The annotation results for seven of the unigene sequences, Comp9719_c0_seq1, Comp9464_c0_seq1, Comp7346_c0_seq1, Comp9464_c0_seq1, Comp7346_c0_seq1, Comp113296_c0_seq1 (105 bp), and Comp9719_c0_seq6 (355 bp), are consistent with the experimental pre-clone known as M. punctipennis sequences in the GenBank database. The annotation results for the Comp9719_c0_seq3, Comp9719_c0_seq4, Comp9719_c0_seq5, Comp64045_c0_seq1 (124 bp) sequences are consistent with the experimental pre-clone known as sequences of Anatolica polita boreali.
Antifreeze proteins
Previous studies have shown that insect AFPs play important roles in cold tolerance, and there are numerous reports that the AFPs are specifically induced in insects that are exposed to low temperatures when they have been shown to improve insect freezing tolerance [40].The M. punctipennis anitfreeze protein (MpAFP) is Cys-, Thr-, and Ser-rich, and ExPASy prediction software indicates that its secondary structure is composed of tandem 12-residue repeats (TCTxSxxCxxAx) with extensive disulfide bond [41, 42]. Three unigenes in our assembly were identified as putatively encoding MpAFP, two of them (Comp9408_c0_seq1 and Comp9408_c0_seq2) have complete ORF. Alignment of the predicted proteins deduced from the two potentially complete unigenes showed that their percentage of identity was 78.19 % (Fig. 5), confirming the remarkable conservation within the AFP family. The relationships among the AFP sequences of M. punctipennies showed that Comp9408_c0_seq1 closed to MpAFPS52, MpAFPS77 and AFP1(Fig. 6). The result could provide the basis for further studies on the function of these genes.

Alignment of the antifreeze protein sequences of M. punctipennis. Identical residues are shaded black, conserved substitutions are shaded grey. Dash (-) indicates insertion or deletion. The antifreeze protein name and GenBank ID of M. punctipennies: MpAFPS52 (ADJ93820.1), AFPS77 (ADJ93819.1), MpAFP1 (AAW67980.1), MpAFP (AAW67979.1)

The homology relationships of M. punctipennies antifreeze proteins. The tree was generated using the neighbor-joining method provided by the software MEGA5 with Poisson correction for multiple amino acid substitutions, and bootstrapping test was performed with 1,000 replicates. The antifreeze protein name and GenBank ID: MpAFPS52 (ADJ93820.1), AFPS77 (ADJ93819.1), MpAFP1 (AAW67980.1), MpAFP (AAW67979.1)
Other candidates
In addition to the unigenes that have been analysed in detail above, other M. punctipennis unigenes with high sequence similarity to important genes related to stress metabolism and targets were identified. In particular, a number of unigenes were annotated as enzymes related to heat or cold metabolic resistance, such as trehalase, trehalose-6-phosphate synthase, chitinase, and cathepsin (Table 4). Although most of these unigenes are not full length sequences, they are nevertheless useful candidates for further characterisation by RACE to retrieve the full length cDNAs. The abundance of these transcripts demonstrates the quality of our sequencing data. This information will provide new leads for functional studies of the genes that play potential roles in beetle resistance to enviroment stress.
Discussion
Reads generation and de novo sequence assembly
The de novo assembly of short reads without a reference genome remains a challenge in spite of the development of many bioinformatics tools for data assembly and analysis [43–46]. Here, we obtained more than 4.8 billion raw reads, and assembled de novo using the Trinity software. We obtained 41,109 unigenes that matched one or more of the searched databases. The unannotated unigenes may represent novel genes whose function has not yet been identified. Specifically, the unigenes had 35,034 (62.17 %) hits to the nr database which was higher than the hits to any of the other databases (Table 2). Most of the top nr matches (first hit) were to sequences from the red flour beetle (T. castaneum) probably because: (1) it demonstrated that M. punctipennis has a near evolution distance with T. castaneum; (2) this is the only beetle with a completely sequenced genome [47]. We mapped more than 15.94 % of the M. punctipennis unigenes to the COG database, 31.97 % to the KOG database, 17.72 % to KEGG pathways and 49.38 % to GO terms, and found that 326 unigenes were homologous to known stress resistance genes. Many other genes and pathways related to stress adaptation were identified but need to be analyzed further.
Heat shock protein genes
HSPs are expressed in most organisms in response to a wide range of stressful environmental conditions and are generally viewed as a protective cellular mechanism [48]. The HSP70 family includes the strictly stress-inducible HSP70 and the constitutive HSC70 (heat shock cognate proteins), the glucose-regulated protein Grp78 (BiP) [49], and the mitochondrial form mitHSP70 (grp75) [50]. In a previous study, we isolated the full length cDNA sequence of a Hsp70 gene from M. punctipennis (Mphsp70) using the RACE-PCR technique. Real-time quantitative PCR showed that the mRNA levels of Mphsp70 at 37 °C and 42 °C was 21.6 and 389.3 fold respectively that of the control at 25 °C, and the mRNA levels decreased as time prolonged at the high temperatures [51]. In the present transcriptome we obtained a considerable number of inducible HSPs genes (72 in total) and we speculated that these genes may help M. punctipennis adapt to the extreme desert environment. Besides, two unigenes (Comp7218_c0_seq1 and Comp7218_c0_seq2) were found similar to the sequence of HSC70 cDNA. Since HSC70 is an important part of the protein folding machinery in a cell [52, 53], the expression of HSC70 in M. punctipennis may help protect its tissues from stress.
Antifreeze protein genes
AFPs were characterized initially in marine fishes [54, 55], where they protect their hosts from freezing by binding to and preventing the growth of seed ice crystals [56]. AFPs lower the freezing point of a solution containing ice below the melting point of the ice. AFPs function both in freeze resistance and freeze avoidance insects, thus AFPs may help insects survive most inhospitable environments.
In previous study, four isoforms of AFPs from M. punctipennis have been isolated and identified [25, 41, 42]. Two of the cDNAs (Mpafps77 and Mpafps52) were from beetles that were collected in summer. The deduced amino acid sequences of the MpAFPs expressed in summer are one 12-residue repeat shorter and have significantly different C-terminal end sequences compared with the MpAFPs expressed in winter [25]. Dozens of AFP isoforms have been indentified in Choristoneura fumiferana [57], Tenebrio molitor [58] and Dendroides canadensis [24]. The function of these AFP isoforms may be different. Six isoforms of cfAFP from C. fumiferana were shown development-specific expression patterns [59]. Similar to C. fumiferana and T. molitor AFPs, the MpAFPs apparently consist of many isoforms with conserved residues [60], which may play important roles in maintaining the integrity of the structure and function of the AFPs. In the present study, three unigene sequences that potentially encode AFPs were identified; their sequences were conserved when aligned with those pre-cloned (Comp9408_c0_seq1 and Comp9408_c0_seq2 vs. MpAFP, MpAFP1, MpAFPS52 and MpAFPS77). This sequences were obtained under different conditions, such as room temperature and cold treatment [25, 41], which suggested that different MpAFPs may have additional functions that were trigered by environmental signals.
Metabolism related unigenes
According to the information provided by GO classification, most of unigenes in the present data were related to metabolism in the biological process. We analyzed 270 unigenes which belong to 13 different groups, and are related to metabolism of M. punctipennis (Table 4). These genes were grouped into the following functions: transmembrane transporter activity (GO:0022857), catalytic activity (GO:0003824), polysaccharide catabolic process (GO:0000272), cysteine-type endopeptidase activity (GO:0004197), etc. Thirteen unigenes were annotated as trehalase (a-glucoside-1-glucohydrolase, EC 3.2.1.28), which is an enzyme that hydrolyzes trehalose to yield two glucose molecules. Trehalose is the major hemolymph sugar in most insects, which acts as an indispensable substrate for energy production and macromolecular biosynthesis [61]. It is predominantly synthesized in the fat body and released into the hemolymph [62]. Trehalase plays a pivotal role in various physiological processes in insect, including flight metabolism [63], chitin synthesis during molting [64], and cold tolerance [65]. All these functions are achieved through the hydrolysis of trehalose (a-D-glucopyranosyl-a-D-glucopyranoside). In the present study, there are 20 unigenes which were annotated as facilitated trehalose transporter (TRET1), and 65 unigenes annotated as chitinase. The expression of these genes in field-collected animals reared under laboratory conditions suggests that M. punctipennis may control the transportation of small molecule to reduce the capacity of the intracellular osmotic potential response to stimuli under non-stress conditions.
According to the information provided by KEGG pathway assignment, 156 unigenes were related to amino sugar and nucleotide sugar metabolism (ko00520) pathway, including chitinase, GDP-mannose 4, 6 dehydratase, hexosaminidase, cytochrome-b5 reductase, UDP-glucose 4-epimerase, etc. The polyhydric alcohols (sorbitol, mannitol, ribitol, erythritol, threitol and ethylene glycol) and sugars (trehalose and glucose) are recognized as cryoprotectant [66]. The discovery of these unigenes may help us to elucidate the metabolic machanism of M. punctipennis that lives in desert environment.
Conclusion
A total of 48,158,004 reads were obtained by transcriptome sequencing and the de novo assembly yielded 56,348 unigenes with an average length of 666 bp. Based on similarity searches with a cut-off E-value of 10−5 against two protein sequence databases, 41,109 unigenes (about 72.96 %) were matched to known proteins. The data presented in this study will contribute significantly to the rapid discovery of a wide diversity of candidate genes for this organism, and will also provide important new insights that will be useful in further studies of M. punctipennis genes and their functions.
References
Cloudsley-Thompson JL (1990) Thermal ecology and behaviour of Physadesmia globosa (Coleoptera: Tenebrionidae) in the Namib Desert. J Arid Environ 19:317–324
Cloudsley-Thompson JL (2001) Thermal and water relations of desert beetles. Naturwissenschaften 88:447–460
Schilmana PE, Kaiserb A, Lighton JRB (2008) Breathe softly, beetle: continuous gas exchange, water loss and the role of the subelytral space in the tenebrionid beetle, Eleodes obscura. J Insect Physiol 54:192–203
Wharton RA (1983) Dispersal, diel periodicity, and longevity of Stips stali (Haag) (Coleoptera: Tenebrionidae). Coleopt Bulletin 37:27–33
Seely MK (1979) Irregular fog as a water source for desert dune beetles. Oecologia 42:213–227
Parker AR, Lawrence CR (2001) Water capture by a desert beetle. Nature 414:33–34
Hamilton WJ III, Henschel JR, Seely MK (2003) Fog collection by Namib Desert beetles. S Afr J Sci 4:181–182
Adam S (2004) Like water off a beetle’s back. Nat Hist 2:26–27
Krasnov B, Ayal Y (1995) Seasonal changes in darkling beetle communities (Coleoptera: Tenebrionidae) in the Ramon erosion cirque Negev Highlands, Israel. J Arid Environ 31:335–347
Wang Y, Liu XN, Zhao Jia, Rexili K, Ma J (2011) The rearing and biology of the desert beetle, Microdera punctipennis, under laboratory conditions. J Insect Sci 11: 39 available online: insectscience.org/11.39
Draney ML (1993) The subelytral cavity of desert tenebrionids. Fla Entomol 76:539–549
Gorb SN (1998) Frictional surfaces of the elytra-to-body arresting mechanism in tenebrionid beetles (Coleoptera: Tenebrionidae): design of co-opted fields of microtrichia and cuticle ultrastructure. Int J Insect Morphol Embryol 27:205–225
Zachariassen KE (1991) Routes of transpiratory water loss in a dry habitat tenebrionid beetle. J Exp Biol Med 157:425–437
Zachariassen KE (1996) The water conserving physiological compromise of desert insects. Eur J Entomol 93:359–367
Storey KB, Storey JM (1988) Freeze tolerance in animals. Physiol Rev 68:27–84
Storey KB, Baust JG, Storey JM (1981) Intermediary metabolism during low temperature acclimation in the overwintering gall fly larva, Eurosta solidaginis. J Comp Physiol 144:183–190
Walters KR, Sformo T, Barnes BM, Duman JG (2009) Freeze tolerance in an Alaska stonefly. J Exp Biol 212:305–312
Irwin JT, Bennett VA, Lee RE (2001) Diapause development in frozen larvae of the goldenrod gall fly, Eurosta solidaginis fitch (diptera: tephritidae). J Comp Physiol B 171:181–188
Rinehart JP, Hayward SAL, Einitsky MA, Sandro LH, Lee RE, Denlinger DL (2006) Continuous up-regulation of heat shock proteins in larvae, but not adults, of a polar insect. Proc Natl Acad Sci USA 103:14223–14227
Salvucci ME, Strecher DS, Henneberry TJ (2000) Heat shock proteins in whiteflies, an insect that accumulates sorbitol in response to heat stress. J Thermal Biol 25:363–371
Parsell DA, Lindquist S (1993) The function of heat-shock proteins in stress tolerance, degradation and reactivation of damaged proteins. Ann Rev Genet 27:437–496
Zachariassen KE, Husby JA (1982) Antifreeze effect of thermal hysteresis agents protects highly supercooled insects. Nature 298:865–867
Hou F, Ma J, Liu X, Wang Y, Liu XN, Zhang FC (2010) Seasonal changes in antifreeze protein gene transcription and water content of beetle Microdera punctipennis (coleoptera: tenebrionidae) from gurbantonggut desert in central Asia. CryoLetters 31:359–370
Duman JG, Verleye D, Li N (2002) Site-specific forms of antifreeze protein in the beetle Dendroides Canadensis. J Comp Physiol 172:547–552
Qiu LM, Mao XF, Hou F, Ma J (2013) A novel function -thermal protective properties of an antifreeze protein from the summer desert beetle Microdera punctipennis. Cryobiology 66:60–68
Huang RX, Wu W, Mao XF, Hu HY, Fan ZT, Hou YJ, Li XP, Du CH, Shao HG, Huang X, Ou YT (2005) The fauna of the desert insects of xiniang and its formation and evolution. Xinjiang Science and Technology Publishing House, XinJiang
Wang L, Si Y, Dedow LK, Shao Y, Liu P, Brutnell TP (2011) A low-cost library construction protocol and data analysis pipeline for Illumina-based strand-specific multiplex RNA-seq. PLoS One 6:e26426. doi:10.1371/journal.pone.0026426
Dunning LT, Dennis AB, Park D, Sinclair BJ, Newcom RD, Buckley TR (2013) Identification of cold-responsive genes in a New Zealand alpine stick insect using RNA-Seq. Comp Biochem Phys D 8:24–31
Zhu JY, Zhao N, Yang B (2012) Global transcriptome profiling of the pine shoot beetle, Tomicus yunnanensis (Coleoptera: Scolytinae). PLoS One 7(2):e32291. doi:10.1371/journal.pone.0032291
Zhang YH, Jiang RX, Wu HS, Liu P, Xie JQ, He YY, Peng H (2012) Next-generation sequencing-based transcriptome analysis of Cryptolaemus montrouzieri under insecticide stress reveals resistance-relevant genes in ladybirds. Genomics 100:35–41
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Fulllength transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652. doi:10.1038/nbt.1883
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res 25:4876–4882
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Consortium GO: gene Ontology: tool for the unification of biology. Nat Genet 25:25–29
Tatusov RL, Koonin EV, Lipman DJ (1997) A genomic perspective on protein families. Science 278:631–637
Wolf YI, Koonin EV (2012) A tight link between orthologs and bidirectional best hits in bacterial and archaeal genomes. Genome Biol Evol 4:1286–1294
Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Rogozin IB, Smirnov S, Sorokin AV, Sverdlov AV, Vasudevan S, IWolf Y, Jodie J Yin JJ, Darren A Natale DA (2004) A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biology 2: Article R7. http://genomebiology.com/2004/5/2/R7
Clark MS, Worland MR (2008) How insects survive the cold: molecular mechanisms–a review. J Comp Physiol B 178:917–933. doi:10.1007/s00360-008-0286-4
Zhao G, Ma J, Xue N, Yang CG, Zhuan FF, Zhang FC (2005) Cloning of a cDNA encoding antifreeze protein in Microdera punctipenis dzunarica (Coleoptera: Tenebrionidae)and its activity assay. Acta Entomol Sin 48:667–673
Qiu LM, Wang Y, Li XM, Ma J (2009) Bioinformatics analysis of antifreeze protein gene Mpafp149 from Microdera punctipennis dzungarica. China J Bioin 7:314–319
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I (2009) ABySS: a parallel assembler for short read sequence data. Genome Res 19:1117–1123
Papanicolaou A, Stierli R, Hffrench-Constant R, Heckel D (2009) Next generation transcriptomes for next generation genomes using est2assembly. Bioinformatics. 10:447. doi:10.1186/1471-2105-10-447
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12:671–682. doi:10.1038/nrg3068
Richards S, Gibbs RA, Weinstock GM, Brown SJ, Denell R, Beeman RW et al (2008) The genome of the model beetle and pest Tribolium castaneum. Nature 452:949–955
Feder ME, Hofmann GE (1999) Heat-shock proteins, molecular chaperones, and stress response: evolutionary and ecological physiology. Ann Rev Physiol 61:243–282
Haas IG (1994) BiP (GRP78), an essential hsp70.resident protein in the endoplasmic reticulum Cmls-Cell. Mol Life S 50:1012–1020
Wadhwa R, Yaguchi T, Hasan MK, Mitsui Y, Reddel RR, Kaul SC (2002) Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res 274:246–253
Ma WJ, Ma J (2012) Cloning and sequence analysis of a heat shock protein gene (Mphsp70) from Microdera punctipennis and its expression related to high temperatures. Chin J Appl Entomol 49:439–447
Dastoor Z, Dreyer J (2000) Nuclear translocation and aggregate formation of heat shock cognate protein 70 (Hsc70) in oxidative stress and apoptosis. J Cell Sci 113:2845–2854
Daugaard M, Rohde M, Jäättelä M (2007) The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710
Devries AL, Wohlschlag DE (1969) Freezing resistance in some Antarctic fishes. Science 163:1073–1075
Duman JG, Devries AL (1974) Freezing resistance in winter flounder Pseudopleuronectes Americanus. Nature 247:237–238
Raymond JA, Devries AL (1977) Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc Natl Acad Sci USA 74:2589–2593
Doucet D, Tyshenko MG, Davies PL, Walker VK (2002) A family of expressed antifreeze protein genes from the moth, Choristoneura fumiferana. Eur J Biochem 269:38–46
Liou YC, Thibault P, Walker VK, Davies PL, Graham LA (1999) A complex family of highly heterogeneous and internally repetitive hyperactive antifreeze proteins from the beetle Tenebrio molitor. Biochemistry 38:11415–11424
Qin W, Doucet D, Tyshenko MG, Walker VK (2007) Transcription of antifreeze protein genes in Choristoneura fumiferana. Insect Mol Biol 16:423–434
Hou XJ, Liang N, Qiu LM, Ma J (2013) Sequence analysis of antifreeze protein genes and their 3′-UTRs from two desert insects, Microdera punctipennis and Anatolica polita. Chin J Appl Entomol 50:1219–1229
Friedman S (1978) Trehalose regulation, one aspect of metabolic homeostasis. Annu Rev Entomol 23:389–407
Kikawada T, Saito A, Kanamori Y, Nakahara Y, Iwata K, Tanaka D, Watanabe M, Okuda T (2007) Trehalose transporter 1, a facilitated and high-capacity trehalose transporter, allows exogenous trehalose uptake into cells. Proc Natl Acad Sci USA 104:11585–11590
Clegg JS, Evans DR (1961) Blood trehalose and flight metabolism in the blowfly. Science 134:54–55
Tatun N, Singtripop T, Sakurai S (2008) Dual control of midgut trehalase activity by 20-hydroxyecdysone and an inhibitory factor in the bamboo borer Omphisa fuscidentalis Hampson. J Insect Physiol 54:351–357
Tatun N, Singtripop T, Tungjitwitayakul J, Sakurai S (2008) Regulation of soluble and membrane-bound trehalase activity and expression of the enzyme in the larval midgut of the bamboo borer Omphisa fuscidentalis. Insect Biochem Molec 38:788–795
Denlinger DL (2010). Low temperature biology of insects
Acknowledgments
This research was supported by the National Natural Science Foundation of China (31360527), and the Open Fund from Xinjiang Key Laboratory of Biological Resources and Genetic Engineering (No. XJDX0201-2014-03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Lu, X., Li, J., Yang, J. et al. De novo transcriptome of the desert beetle Microdera punctipennis (Coleoptera: Tenebrionidae) using illumina RNA-seq technology. Mol Biol Rep 41, 7293–7303 (2014). https://doi.org/10.1007/s11033-014-3615-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-014-3615-6