
Abstract
This paper presents an approach to semi-automated data analysis, supported by tools for pattern construction, exploration and explanation. The proposed three-part methodology for multiresolution 0–1 data analysis consists of data clustering with mixture models, extraction of rules from clusters, as well as data and rule visualization using banded matrices. The results of the three-part process: clusters, rules from clusters, and banded structure of the data matrix are finally merged in a unified visual banded matrix display. The incorporation of multiresolution data is enabled by the supporting ontology, describing the relationships between the different resolutions, which is used as background knowledge in the semantic pattern mining process of descriptive rule induction. The presented experimental use case highlights the usefulness of the proposed methodology for analyzing complex DNA copy number amplification data, studied in previous research, for which we provide new insights in terms of induced semantic patterns and cluster/pattern visualization. The methodology is successfully evaluated on four other publicly available data sets, which further demonstrates the utility of the proposed approach.
Similar content being viewed by others


1 Introduction
Data analysis is concerned with finding ways to summarize the data to become easily understandable (Hand et al. 2001). The interpretation aspect is especially valued among domain specialists who may not understand the data analysis process itself. In the current age of big data and accompanying complex models, understandability and interpretability of the models is even more essential as, according to Richard Hamming, “the purpose of computing is insight, not numbers” (Hamming 1986). For complex models generated from big data, understanding these models will help to understand the data generating phenomenon and help to make better decisions based on the data (Kuhn and Johnson 2013). Semi-automated data analysis is hence made possible for the end-user if data analysis processes are supported by easily accessible methodologies and tools for pattern and model construction, as well as their exploration and explanation.
This work combines different approaches developed in our previous research, leading to a new three-part data analysis methodology, whose utility is demonstrated in a case study concerning the analysis of DNA copy number amplifications represented as a 0–1 (binary) data set (Myllykangas et al. 2006). In previous work, we have successfully clustered this data using mixture models (Myllykangas et al. 2008; Tikka et al. 2007). Furthermore, in Hollmén and Tikka (2007), we have learned linguistic names for the patterns that coincide with the natural structure in the data, enabling domain experts to use these names to refer to the clusters or to the patterns extracted from the clusters. In Hollmén et al. (2003) we reported that frequent itemsets describing the clusters, or extracted from the ‘one cluster at a time’ clustered data differ from those extracted from the whole data set. The whole set of about 100 DNA amplification patterns identified from the data have been described in Myllykangas et al. (2008).
In the proposed approach we start from our initial studies of using mixture models to crossover unsupervised methods of probabilistic clustering with supervised methods of subgroup discovery with the aim to determine the chromosomal locations that are responsible for specific types of cancers. We also enrich the data with additional background knowledge that enables the analysis of data at multiple resolution levels. Specifically, with the aim of better explaining the initial mixture model based clusters, the proposed methodology considers the cluster identifiers as class labels for descriptive rule learning (Novak et al. 2009), using semantic pattern mining (Vavpetič et al. 2014). The resulting semantic rules are generated by the Hedwig semantic pattern mining algorithm (Vavpetič et al. 2013) performing semantic subgroup discovery by using the incorporated background knowledge in the form of pre-discovered patterns as well as taxonomies of features in multiresolution data. Finally, we use a banded matrix approach to visualize the clustering result and rules obtained from semantic subgroup discovery overlayed on the same data, thus providing holistic picture of the data and consequently, of the data generating phenomenon.
Explaining the obtained clustering results to the users is essential. It was shown that in text mining (Hotho et al. 2003), semantic structures can be used to explain the clustering results at an appropriate level of granularity. Similarly, a methodology consisting of clustering and semantic pattern mining, has already been suggested in our previous work (Langohr et al. 2013; Vavpetič et al. 2014). However, in this work we have for the first time addressed the task of explaining sub-symbolic mixture model patterns (clusters of instances) using symbolic rules. To this end, we propose our previous approach (Vavpetič et al. 2014) to be enhanced through pattern comparison by their visualization on the plots resulting from banded matrices visualization (Garriga et al. 2011). Using different color schemes on the banded matrix structure (induced from the original data), the mixture model clusters are first visualized, followed by visualizing the sets of patterns (i.e. subgroups) induced by semantic pattern mining. The proposed visualization provides new means for data and pattern exploration and comparison. To the best of our knowledge, such a three-part exploratory approach to data analysis has not been proposed in the data mining literature before.
The main contribution of this work is a three-part methodology for data analysis, consisting of (i) data clustering, (ii) extraction of semantic patterns (rules) from the clusters, using an ontology of relationships between the different resolutions of the multiresolution data, and (iii) integration of the results in a visual display, illustrating the clusters and the identified rules by visualizing them over the banded matrix structure, first described in Adhikari et al. (2014). This work significantly extends our previous report on the same topic (Adhikari et al. 2014) in many ways. First of all, we used a more elaborate experimental setting with four additional data sets. Furthermore, we added a new section on literature survey where we present the state-of-the-art in all three methodological parts in our contribution as well as the holistic picture of similar methodologies, and sections detailing the model selection procedure in mixture models and performed statistical tests for empirical verification of stability of the clustering results. We also changed a part of the methodology, replacing one banded matrix algorithm (the barycentric method) with another (the bidirectional MBA) which yielded better results in our experiments.
The paper is structured as follows. The related work is presented in Sect. 2. Methodology overview along with the details are explained in Sect. 3. Section 4 describes the experimental data sets. Section 5 describes the experiments on the chromosomal amplification data set and their results, while Sect. 6 presents the experiments on four additional publicly available data sets. We present the results of the stability analysis of clustering results in Sect. 7. In Sect. 8, we summarize the results and conclude the paper.
2 Related work
The following sections provide a brief overview of related work in mixture modeling, analysis of multiresolution data, semantic pattern mining, and pattern visualization using banded matrices. In the end of this section, we review some of the research that investigates at least two aspects of our three-part methodology.
2.1 Mixture models
Mixture models have been popular in the probabilistic modeling domain because of their flexibility in the choice of component distributions and their applicability to a wide variety of applications. Mixture models are at the heart of model based clustering (Melnykov and Maitra 2010). Authors in Melnykov and Maitra (2010) review the model based clustering approach in different application areas, such as text mining, proteomics, and medical data analysis. Similarly, authors in McLachlan and Peel (2000) summarize different application areas where mixture models have been used with plausible results such as density estimation, missing data imputation, combining different density models, and model heterogeneity. In our earlier work, mixture models were used to model heterogeneous cancer patient data (Myllykangas et al. 2008; Tikka et al. 2007).
2.2 Mixture models in copy number analysis
In the beginning, DNA copy number analysis focused in determining the copy number of the cytogenetic bands (Knuutila et al. 1999; Pollack et al. 1999). However, in Knuutila et al. (1999) and Pollack et al. (1999), the authors did not establish a relation between the copy numbers and their clinical significance.
DNA copy number amplification data collected from bibliomics survey from 838 journal articles published from 1992 to 2002 was analyzed in Myllykangas et al. (2006), where amplification patterns were determined for 73 different neoplasms and the neoplasms were clustered according to amplification profiles thus identifying the amplification hotspots using independent component analysis. The profiling revealed that human neoplasms formed clusters based on the amplification frequency of the cancer. Similarly, authors in Myllykangas et al. (2008) classified the human cancers based on copy number amplification using probabilistic modeling. Furthermore, the authors extracted the ranges of the amplification in the chromosome and expressed it according to the cytogentic nomenclature.
In Hollmén and Tikka (2007) and Tikka et al. (2007), the authors modeled the DNA copy number amplifications using a mixture of multivariate Bernoulli Distributions. The classification of 73 different neoplasms in Myllykangas et al. (2006) were extended to 95 different neoplasm types. Furthermore, in Rancoita et al. (2009), the authors have proposed the enhancement to Bayesian Piecewise Constant Regression (BPCR), called mBPCR, changing the segment number estimator and boundary estimator to enhance the fitting procedure. The proposed mBPCR was more accurate in determining the true breakpoints of amplification. More recent studies Despierre et al. (2010) and D’haene et al. (2010) have mainly focused in cancer specific analysis of DNA copy number.
2.3 Multiresolution data analysis
Multiresolution data arises when a phenomenon is measured with varying precision (Willsky 2002). A phenomenon measured with increasing precision measures the finer details of the phenomenon and produces the data in fine resolution. In contrast, a phenomenon measured with decreasing precision measures the coarser details of the phenomenon and produces data in coarse resolution. Multiresolution data are abundant in domains such as time series, image processing, geoinformatics, and telecommunications (Willsky 2002). Multiresolution methods are gaining popularity in recent years because of their ability to model data in multiple dimensions within a single analysis, providing means to combine multiple data sets and sources within a single analysis framework.
Multiresolution modeling is closely related to the scale space theory (Lindeberg 1994) and multiscale analysis (Weinan 2011) and the terms are sometimes used interchangeably in the literature. Multiscale representation is often generated from single resolution data by successive smoothing and subsampling, for example, by using the pyramid structure in image processing domain (Lindeberg 1994). Scale space representation improves over multiscale representation by providing facilities to compute representation using a desired scale parameter, t. Scale space and multiscale methods work in the model domain where models represent single resolution data at different scales. In contrast, multiresolution modeling problem arises in the data domain where the same data generating system is measured at varying levels of detail. Wavelets describe mathematical phenomena, such as functions and signals at different levels of resolution but in a regular, consistent and homogeneous setting (Jawerth and Sweldens 1994). Most of propositional machine learning and data mining methods described in the literature are designed to work with single resolution data. Since the dimensionality of different data resolutions is different, the usual approach is to model each resolution separately. Scale space methods and wavelets usually use a multiresolution analysis setting for the data sets in the same resolution. Furthermore, the multiresolution scenarios where wavelets and scale space methods have their usage require regular, consistent, and homogeneous division of regions, such as the pyramid structure in the image processing domain (Wilson 2000). In a multiresolution setting, the division is consistent but irregular because a region in a coarse resolution is not always divided into the same number of regions in a fine resolution like in our multiresolution chromosomal amplification data sets.
Multiresolution mixture models have been proposed in the literature. For example, a multiresolution Gaussian mixture model founded on the pyramid structure in image processing domain models the visual motion in Wilson (2000). Authors in Mukherjee et al. (2013) incorporate wavelet sub-bands in a Gaussian mixture model to improve their performance thereby providing a generic platform to use any multiresolution decomposition based Gaussian mixture model for background suppression. We adapted mixture modeling for multiresolution data in our past research. In Adhikari and Hollmén (2010), we transformed the multiresolution data to a single resolution and applied the mixture modeling algorithm on the combined data thus increasing the performance of mixture models on single resolution data. In Adhikari and Hollmén (2013), we showed the improvement in the modeling performance of multiresolution mixture model by designing the structure of multiresolution components from the domain knowledge for the mixture model such that a single multiresolution component is a Bayesian network.
2.4 Semantic pattern mining
Rule learning, which was initially focused on building predictive models formed of sets of classification rules, has recently shifted its focus to descriptive pattern mining. Well-known pattern mining techniques are based on association rule learning (Agrawal and Srikant 1994; Piatetsky-Shapiro 1991). While the initial studies in association rule mining have focused on finding interesting patterns from large data sets in an unsupervised setting, association rules have been used also in a supervised setting, to learn pattern descriptions from class-labeled data (Liu et al. 1998). Building on top of the research in classification and association rule learning, subgroup discovery has emerged as a popular data mining methodology for finding patterns in class-labeled data. Subgroup discovery aims at finding interesting patterns as individual rules that best describe the target variable (Klösgen 1996; Wrobel 1997).
Subgroup descriptions in the form of propositional rules are suitable descriptions of groups of instances. However, given the abundance of taxonomies and ontologies that are readily available, these can also be used to provide higher-level descriptors and explanations of discovered subgroups. Especially in the domain of systems biology, the GO ontology (Gene Ontology Consortium 2008), KEGG orthology (Ogata et al. 1999) and Entrez gene–gene interaction data (Maglott et al. 2005) are good examples of structured domain knowledge that can be used as additional higher-level descriptors in the induced rules.
The challenge of incorporating domain ontologies in data mining was addressed in recent research on semantic data mining (SDM) (Lawrynowicz and Potoniec 2011; Vavpetič and Lavrač 2013). Using ontologies, authors in Lawrynowicz and Potoniec (2011) introduce an algorithm named Fr–ONT for frequent concept mining expressed in \(\mathcal {EL}^{++}\) DL. In Vavpetič and Lavrač (2013), we described and evaluated the SDM toolkit that includes two semantic data mining systems: SDM-SEGS and SDM-Aleph. SDM-SEGS is an extension of the earlier domain-specific algorithm SEGS (Trajkovski et al. 2008) which allows the application of semantic subgroup discovery in gene expression data. SEGS constructs gene sets as combinations of GO ontology (Gene Ontology Consortium 2008) terms, KEGG orthology (Ogata et al. 1999) terms, and terms describing gene–gene interactions obtained from the Entrez database (Maglott et al. 2005). SDM-SEGS extends and generalizes this approach by allowing the user to input any set of ontologies in the OWL ontology specification language and an empirical data collection which is annotated by domain ontology terms. SDM-SEGS employs ontologies to constrain and guide the top-down search of a hierarchically structured space of induced hypotheses. SDM-Aleph, which is built using the inductive logic programming system Aleph (Srinivasan 2007), does not have the limitations of SDM-SEGS, imposed by the domain-specific algorithm SEGS. Additionally, SDM-Aleph can accept any number of OWL ontologies as background knowledge, which are then used in the learning process.
Based on the lessons learned in Vavpetič and Lavrač (2013), we introduced a new system Hedwig in Vavpetič et al. (2013). The system takes the best from both SDM-SEGS and SDM-Aleph. It uses an efficient search mechanism tailored to exploit the hierarchical nature of ontologies. Furthermore, Hedwig can take into account background knowledge in the form of RDF triplets. Compared to Vavpetič et al. (2013), we upgraded the original system to use better redundancy pruning and significance tests based on Hämäläinen (2010). The latest version of Hedwig supports also negations of unary predicates. This version of the Hedwig system was used in the experiments described in this paper.
2.5 Related methodologies
Complex models are needed for modeling complex, non-linear relationships in the data. As argued in Thrun (1995), however, complex models exhibit a low degree of human comprehensibility. Rules can be used to represent complex models, since they have the advantage of being compact, modular, explicit and interpretable by domain experts (Tresp et al. 1997). In our current work, we use semantic pattern mining to represent the clustered data in an interpretable fashion. Another line of work is to summarize the clustered data in an interpretable fashion in the context of topic models (Mei et al. 2007; Lau et al. 2011). Having identified topics as clusters in a document collection, the task is to summarize the contents of that cluster or topic in a concise way.
Work presented in Tresp et al. (1997) considers relationships between probabilistic rules, normalized Gaussian basis functions and Gaussian mixture models, which can be seen as different representational forms of knowledge. The work considers extracting rules out of models, but also the use of rules to support model estimation. Rule extraction from feed-forward neural networks is investigated in Thrun (1995). In that work, rules are extracted, where the precondition is given by a set of intervals for the individual values and the output is a single target category.
The aim of the research presented in Lau et al. (2011) is to automatically generate topic labels which explicitly identify the semantics of the topic. The work in Mei et al. (2007) proposes probabilistic approaches to automatically labeling multinomial topic models in an objective way.
2.6 Data clustering and visualization using banded matrices
Data visualization has been an integral ingredient in the overall data mining process because it presents insights into complex data sets by communicating their key aspects (Tufte 1986). Furthermore, providing information in the visual format is one of the fastest and best methods understandable to domain experts. Data is often represented in a matrix form, and research community has developed numerous methods for matrix visualization (Chen et al. 2004; Wu et al. 2010). In this contribution, we use banded matrices to visualize the data and the results of a data mining process in a way that the results become easily understandable to the domain specialist.
While binary matrices are frequently used as input in data mining (perhaps the most notable example of binary matrices being market basket data), the concept of banded matrices has its origins in numerical analysis. This is because the computational effort of multiplying matrices is much smaller when matrices are banded. The interest of the numerical community is usually in reducing the total bandwidth of a matrix. This differs slightly from the interests in data mining, where the goal is to find a matrix structure as close to a banded one with the underlying assumption that the data analyzed is noisy and contains outliers. The connection between banded matrices and their relation to data analysis was initially studied in Garriga et al. (2011), where several algorithms were proposed to find optimal permutations of rows (and sometimes columns) that best expose the banded structure of a matrix.
In this work, we conducted experiments with three algorithms: minimal banded augmentation (MBA), bidirectional MBA (biMBA), and the barycentric method. Given that the performance of the biMBA method, first proposed in Garriga et al. (2011), was superior to both MBA and the barycentric method, we used this method in the visualization.
3 Methodology
This section describes the proposed three-part methodology of our contribution. The three steps consist of clustering with mixture models, a subsequent cluster explanation through pattern construction using semantic pattern mining, and finally pattern visualization enabling improved pattern interpretation.
3.1 Methodology overview
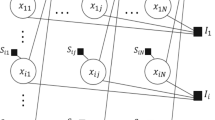
The proposed methodology is illustrated in Fig. 1. The input to the methodology pipeline is the experimental data and the background knowledge, which defines the taxonomy of attribute values at different levels of the given multi-resolution data, with locations for various factors that are known to contribute to cancer development or are characteristic of most cancer types.
The first step in the methodology pipeline is mixture modeling, consisting of model selection to determine the number of mixture components and probabilistic clustering to generate the cluster labels from the data. In the next step, data is structured using a banded matrix approach. While the banded structure is induced from the data independently of cluster labels and the background knowledge, the obtained banded structure can be used also to support the visualization of the clusters obtained through mixture modeling. Next, the data (labeled by cluster labels obtained from mixture modeling) and the background knowledge are used as input to the Hedwig semantic pattern mining algorithm, to get the descriptions of data clusters in the form of logical rules, whose conditions include conjunctions of background knowledge concepts. Semantic pattern mining is the only modeling approach in the methodology that uses the background knowledge and facts. Finally, all three models (the mixture model, the banded matrix and the patterns) are joined to produce the final banded matrix-based visualization.

Overview of the proposed three-part methodology used in the analysis of high-dimensional multiresolution data
3.2 Mixture model clustering
Mixture models are probabilistic models for modeling complex distributions by a weighted sum, or a mixture of simple distributions. Mixture model decomposes the complex probability distribution into a set of component distributions (McLachlan and Peel 2000). The form of mixture distribution is dependent on the choice of the component distributions. Distributions from exponential family such as Gaussian and Dirichlet dominate the choice of component distributions (McLachlan and Peel 2000). Since the data set of our interest is a 0–1 data, we use multivariate Bernoulli distributions as component distributions to model the data. Mathematically, this can be expressed as:
Here, \(j=1,2, \ldots , J\) indexes the component distributions and \(i=1,2, \ldots , d\) indexes the dimensionality of the data. \(\pi _j\) defines the mixing proportions or mixing coefficients determining the weight for each of the J component distributions. The mixing coefficients satisfy the properties of convex combination, i.e., \(\pi _{j} \ge 0 \) and \(\sum _{j=1}^{J} \pi _{j}=1\). Individual parameters \(\theta _{ji}\) determine the probability that a random variable in the \(j^{th}\) component in the \(i^{th}\) dimension takes the value 1. Parameters for a component distribution j is denoted as \(\varvec{\theta }_j=(\theta _{j1},\theta _{j2}, \ldots , \theta _{jd})\). The term \(x_i\) denotes the data point such that \(x_i \in \{0,1 \} \), in the data vector \(\mathbf {x} = (x_1,x_2,\ldots x_d)\). Therefore, the parameters of mixture models can be represented as: \(\varvec{\varTheta }=\{J\), \(\{\pi _j, \varvec{\theta }_j \}_{j=1}^{J}\}\). We can formulate Eq. 1 in log-likelihood terms according to maximum likelihood principle (Bishop 2006), where parameter values that maximise the log-likelihood can be defined as:
3.2.1 Motivation behind using mixture models
Whereas the mixture model is merely a way to represent the probability distribution of the data, the model can be used in clustering the data into (hard) partitions, or subsets of data instances. We can achieve this by allocating individual data vectors to mixture model components that maximize the posterior probability of that data vector.
Among the diverse set of clustering methods of choice we chose mixture modeling because we wanted to model the data in a probabilistic context. Probabilistic models used in clustering provides several advantages over traditional clustering methods as they provide principled methods to address issues such as number of clusters, and missing variables (McLachlan and Peel 2000). Clustering methods such as k-means (which can also be interpreted as mixture models) use simple statistical measures such as mean, or median of data items in clusters, while we opted for mixture models that provide more complete information. When mixture models are used in clustering, the components represent the clusters making it possible to obtain density estimation for each cluster (Bishop 2006). Similarly, mixture model covers the data well as the dominant patterns are captured by the components of the mixture model. A mixture model with high likelihood results in component distributions with high peaks, which means that the data in clusters are dense (Kononenko and Kukar 2007).
Traditional clustering algorithms such as k-means utilize unsupervised learning to group samples that are ‘near’ each other according to predefined measure of similarity (Jain et al. 1999). These methods are more suitable for continuous data which has well defined distance measures. Although several similarity measures are defined for binary data, their application in binary data is not straightforward. Furthermore, our major application area was cancer genetics and cancer is not a single disease but a heterogeneous collection of several diseases. Mixture models are well-known for their ability to model heterogeneity (McLachlan and Peel 2000). In the current application we have used unsupervised clustering on cancer data sets with multiple cancer types, hence, one cluster can contain cancer types from multiple cancers. Mixture models also provide the facility of soft clustering, however, soft clustering is out of the scope of this work.
3.2.2 Model selection in mixture models
Expectation Maximization (EM) algorithm can be used to learn the maximum likelihood parameters of the mixture model if the number of component distributions are known in advance (Dempster et al. 1977). However, the number of components (i.e. number of clusters) in the data is often unknown a priori in most real-world applications. Hence, model selection is also an essential prerequisite of learning mixture models. Model selection is the process of choosing a model of appropriate complexity that fits the given data set optimally (Cherkassky and Mulier 1998; Hastie et al. 2009). The complexity parameter in mixture model is the number of mixture components, therefore, model selection in mixture model is the choice of appropriate number of components in the mixture model.
A plethora of criteria have been proposed in the literature to determine the appropriate number of mixture model components (McLachlan and Peel 2000). For example, authors in Celeux (2007), Figueiredo and Jain (2002), and Oliveira-Brochado and Martins (2005) comprehensively review deterministic, stochastic and resampling criteria to evaluate the performance of mixture model and therefore select the model of appropriate complexity. Deterministic criteria consists of Akaike Information Criterion (AIC), Bayesian Information Criterion (BIC), Minimum Description Length (MDL), and integrated classification likelihood (ICL). Similarly, stochastic methods include Markov Chain Monte Carlo (MCMC), and resampling methods include bootstrapped likelihood ratio test (McLachlan 1987), while authors in Woo and Sriram (2006) propose a robust approach against model mis-specification leading to a better fitting mixture density based on minimum Hellinger distances. In addition, the authors in Chen and Khalili (2008) and Huang et al. (2013) use penalised likelihood method for model selection in mixture model.
A popular criterion measure of the quality of mixture models is the data likelihood (Smyth 2000). In addition, cross-validation is widely used model validation technique. Therefore, we use cross-validated likelihood to select the model of appropriate complexity as documented in Tikka et al. (2007). A mixture model with large number of mixture components produces larger value for the log-likelihood in Eq. 2 for training data. However, a mixture model with large number of mixture components also overfits the data, and generalizes poorly on the future unseen data. Additionally, mixture models with large number of components require greater resources: both time and memory. In contrast, a mixture model with smaller number of mixture components results in an underfitted model, and is unable to adequately represent the underlying true data distribution. Therefore, model selection aims to optimize this trade-off between too simple and too complex models (McLachlan and Peel 2000). A well trained mixture model with appropriate number of mixture components estimates the underlying data distribution better and produces high likelihood values for the unseen data which is the primary objective of our model selection procedure (Bishop 2006).
3.3 Semantic pattern mining
The expansion of the semantic web and increasing availability of domain knowledge in the form of ontologies have resulted in the growth of semantic data. Consequently, ontologies are recognized as useful for encoding semantics of data also in the machine learning and data mining communities and recent studies have shown that additional knowledge can enhance the knowledge discovery process (Panov 2012). Note that—in contrast to the philosophical definition of ontology—we use the plural form ontologies to emphasize that they can be independent domain models, possibly obtained from different sources.
In our application area, ontologies come from known biological landmarks or other known biological information. Similarly, many application areas have readily available background information that could prove useful in the data analysis process, especially in biological and clinical applications. Semantic data mining addresses this challenge of mining the abundance of available knowledge encoded in domain ontologies to improve the process of data mining (Vavpetič et al. 2014).
Existing semantic subgroup discovery algorithms are either specialized for a specific domain (Trajkovski et al. 2008) or adapted from systems that do not take into the account the hierarchical structure of background knowledge (Vavpetič and Lavrač 2013). On the other hand, recently developed semantic subgroup discovery system Hedwig (Vavpetič et al. 2013), is designed as a general purpose semantic subgroup discovery system that uses domain ontologies to structure the search space to formulate the hypotheses using ontology concepts.
Semantic subgroup discovery, as addressed by the Hedwig system, results in relational descriptive rules. Hedwig uses ontologies as background knowledge and training examples in the form of Resource Description Framework (RDF) triples. Formally, we define the semantic data mining task addressed in this work as follows.
Given:
-
the empirical data in the form of a set of training examples expressed as RDF triples,
-
domain knowledge in the form of ontologies, and
-
an object-to-ontology mapping which associates each object from the RDF triplets with appropriate ontological concepts.
Find:
-
a hypothesis (a predictive model or a set of descriptive patterns), expressed by domain ontology terms, explaining the given empirical data.


Subgroup describing rules are first-order logical expressions. Consider the following rule used to explain the format of induced subgroup describing rules, for example:

with True Positives (TP)=80 and False Positives (FP)=20. Variables X, Y represent sets of input instances, R is a binary relation between the examples and \(C_1, C_2\) are ontological concepts. This rule is interpreted as follows: if an example X is annotated with concept \(C_1\), and is related with an example Y via R, and Y is annotated with concept \(C_2\), then the conclusion Class(X) holds. This rule condition is true for 100 input instances (\(TP+FP\), also called coverage), 80 of which are of the target class (TP, also called support).
The Hedwig system, which implements Algorithms 1 and 2 to search for interesting subgroups, supports ontologies and examples to be loaded as a collection of RDF triples (a graph). The system automatically parses the RDF graph for the subClassOf hierarchy, as well as any other user-defined binary relations. Hedwig also defines a namespace of classes and relations for specifying the training examples to which the input must adhere.
The algorithm uses beam search, where the beam contains the best N rules found so far. The search starts with the default rule which covers all the input examples. In every iteration of the search, each rule from the beam is specialized via one of the four operations:
-
1.
Replace predicate of a rule with a predicate that is a sub-class of the previous one,
-
2.
Negate predicate of a rule,
-
3.
Append a new unary predicate to the rule,
-
4.
Append a new binary predicate, thus introducing a new existentially quantified variable (note that the new variable needs to be ‘consumed’ by a literal to be conjunctively added to this clause in the next step of rule refinement).
Rule induction via specializations is a well-established way of inducing rules, since every specialization either maintains or reduces the current number of covered examples. A rule will not be specialized once its coverage is zero or falls below some predetermined threshold. When adding a new conjunction, we check that if the extended rule does not improve the probability of the conclusion (we use the redundancy coefficient, as in Hämäläinen 2010), then it is not added to the pool of specializations. After the specialization step is applied to each rule in the beam, we select new set of the best scoring N rules. If no improvement is made to the collection of rules, the search is stopped. In principle, our procedure supports any rule scoring function. Numerous rule scoring functions (for discrete targets) are available: \(\chi ^2\), precision, WRAcc (Lavrač et al. 2004), leverage and lift. The latter is the default choice and was also used in our experiments. After the induction phase, the significance of the findings is tested using the Fisher’s exact test (Fisher 1922). To cope with the multiple-hypothesis testing problem, we use Holm-Bonferroni (Holm 1979) direct adjustment method with \(\alpha =0.05\).
3.4 Visualization using banded matrices
Consider a binary matrix M with N rows and d columns and two permutations, \(\kappa \) and \(\pi \) of the first N and d integers. Matrix \(M_{\kappa }^{\pi }\), defined as \(\left( M_{\kappa }^{\pi }\right) _{i,j} = M_{\kappa (i), \pi (j)}\), is constructed by applying the permutations \(\pi \) and \(\kappa \) on the rows and columns of M. If, for some pair of permutations \(\pi \) and \(\kappa \), matrix \(M_{\kappa }^{\pi }\) has the following property:
-
Each row i of the matrix has the consecutive ones property. This means that the column indices for which the value in the matrix is 1 appear consecutively, i.e. on indices \(a_i, a_i+1, \ldots , b_i\),
-
For each i, we have \(a_i \le a_{i+1}\) and \(b_i \le b_{i+1}\),
then the matrix M is fully banded. Furthermore, if matrix M is fully banded, then its transpose \(M^{\top }\) is also fully banded.
Figure 2 demonstrates the motivation behind banded matrices as it shows that finding the banded structure of a matrix simultaneously exposes the clustered structure of the underlying data. This means that banded matrix factorization can provide an evaluation of the clustering results—we expect that clusters, discovered in a data set, will also be exposed by the banded matrix visualization. Similar visual perspective can also be shown by displaying all the clusters together, however, using independent banded matrices on them gives more validity to the results. Allowing the samples from the same cluster to spread along the matrix will ease pattern comparison as similar patterns from different clusters will be grouped together. Additionally, it is easier to see the similar clusters in the data and make future decisions such as splitting of clusters or merging of clusters for future experiments. When the reordering selected does not depend on the cluster structure discovered, the resulting figures offer new insight into both the data and the clustering.
For a fully banded matrix, it can be shown that a banded structure can be found in polynomial time (Garriga et al. 2011). We cannot expect, however, that real world matrices, especially those originating in a disease as heterogeneous as cancer, will be fully banded. The problem is that, for a matrix involving noise, finding the correct row and column permutations that show a structure, close to a banded one, may be computationally unfeasible. We, therefore, need algorithms that attempt not only to find column and row permutations that are as close to banded as possible in some sense, but also find these ‘almost banded’ structures in a decent time frame.

An example of a binary matrix before and after row and column permutations exposing a banded structure
The method used to find the banded structure of a matrix in this article, called the bidirectional minimum matrix augmentation (biMBA) method, was first proposed in Sugiyama et al. (1981) and was first used as a method of banded matrix extraction in Garriga et al. (2011). One step of the method consists of three substeps,
3.4.1 Ensuring the maximum ones property
In the first step, each row of the matrix is transformed to have the consecutive ones property by finding the smallest number of matrix elements that have to be changed (either from 1 to 0 or from 0 to 1) for the row to have the consecutive ones property.
Theorem 1
Given a matrix M, finding the correct elements to change in the i-th row of M is equivalent to solving the maximum subarray problem for the matrix W, defined as
Proof
The transformation of the matrix row i into one with a consecutive ones property is obviously an operation that results in the row having elements \(a, a+1,\ldots , b\) set to 1 and the remaining elements set to 0 (for some pair of integers \(1\le a\le b\le n\)), so the task of finding this transformation is equivalent to finding the correct (those that require the smallest number of matrix element changes) values for a and b. The number of matrix element changes assigned to each value of (a, b) is equal to
The task of finding the smallest number of matrix element changes to make the row have the consecutive ones property is therefore equivalent to finding \(\displaystyle \mathrm {argmin}_{a\le b} C_i(a,b)\)
On the other hand, solving the maximum subarray problem for the i-th row of matrix W is defined as finding the subarray of the matrix for which the sum of the elements is the biggest. Just as before, each subarray can be represented by two integers a, b which represent the start and end point of the subarray. The maximum subarray problem is equivalent to finding \(\mathrm {argmax}_{a\le b} P_i(a,b)\), where \(P_i\) is defined as \(P_i(a,b) = \sum _{i=a}^b W_{i,j}\). We know that the elements of W can only equal 1 or \(-1\), so \(P_i(a,b)\) can be rewritten as
which can, following the definition of \(W_{i,j}\), be written as
We now consider the fact that the set \(S_i =\{j| M_{i,j} = 1\}\) (which is fixed for a given i) is the disjoint union of the sets \(S_{i, \in } = \{a\le j\le b| M_{i,j} = 1\}\) and \(S_{i,\notin } = \{(j<a\vee j>b)\wedge M_{i,j} = 1\}\) and we see (since \(|S_i| = |S_{i,\in }| + |S_{i,\notin }|\)) that
This shows that \(P_i(a,b) = \mathrm {const.} - C_i(a,b)\), meaning that \(\mathrm {argmin}(C_i) = \mathrm {argmax}(P_i)\), concluding the proof.
Theorem 1 shows that for each row i, the elements which have to be changed to transform it into a consecutive ones row can be found by solving a maximum subarray problem which is solvable in linear time by finding, for each index j, the best subarray \(s_j\) ending at j. If \(W_{i,j}=-1\), then \(s_j\) is obviously equal \(s_{j-1}\) with the addition of j (if the sum of the elements of \(s_{j-1}\) is positive) or it is an empty array with sum 0 (if the sum of the elements of \(s_{j-1}\) is zero). On the other hand, if \(W_{i,j} = 1\), then adding j to the subarray \(s_{j-1}\) clearly makes the best possible subarray ending at j.
After transforming M into a matrix with the consecutive ones property, we denote the new matrix \(M'\).
3.4.2 Ensuring the existence of a banded structure
To ensure the existence of a banded structure for \(M'\), we now must further ensure that there is no pair of rows \(i_1, i_2\) such that \(a_{i_1}< a_{i_2}\) and \(b_{i_2}<b_{i_1}\) (the interval of ones in row \(i_2\) is *completely subsumed* by the interval of ones in row \(i_1\)). It is obvious that if such a pair exists, then \(M'\) is not fully banded, since according to \(a_{i_1}, a_{i_2}\), the row \(i_1\) should be above row \(i_2\) ,but according to \(b_{i_1}, b_{i_2}\), the row \(i_2\) should be above row \(i_1\). However, as shown in Garriga et al. (2011), the reverse also holds: if no such pair \(i_1, i_2\) exists, then the matrix is fully banded.
This can be seen since if no such pair exists, we can sort the matrix rows by \(a_i\), then by \(b_i\) to obtain a fully banded matrix \(M''\). For any row i of \(M''\) (with consecutive ones between \(a_i\) and \(b_i\)), we then know that if \(a_i=a_{i+1}\), we will have \(b_i\le b_{i+1}\) by our sorting, and if \(a_i < a_{i+1}\), then \(b_i>b_{i+1}\) would mean that before sorting, row \(i+1\) had an interval of ones that was completely subsumed by the interval of ones in row i, which is not possible.
In order to eliminate fully subsumed pairs of rows, in the second step, the algorithm finds each pair of rows \(i_1, i_2\) such that \(a_{i_1}< a_{i_2}\) and \(b_{i_1}> b_{i_2}\). Then, for each such pair, the algorithm performs the minimum number of matrix element changes required so that either \(a_{i_1} = a_{i_2}\) (this is done by adding ones before \(a_{i_2}\) to row \(i_2\)) or \(b_1=b_2\) (by adding ones after \(b_{i_2}\) to row \(i_2\)) or by completely deleting all ones in row \(i_2\). Because all changes are made to row \(i_2\), if we traverse the pairs \(i_1,i_2\) in a double for loop, we can be sure that no completely subsumed intervals will be created anew, meaning that the result of this step is a fully banded matrix.
3.4.3 Finding the permutation to show the banded structure of \(M''\)
As we have shown in the previous two points, the matrix \(M''\) is fully banded. Furthermore, there exists a permutation \(\pi \) of the rows of \(M''\) that exposes the banded structure of \(M''\). This permutation can be found by simply sorting the starting points of the intervals of ones in the rows of \(M''\) from smallest to largest, resolving ties by the endpoints of the intervals (sorting first by \(a_i\), then by \(b_i\)).
Following the steps outlined above, Algorithm 3 calculates the best possible (in some way) permutations of rows that will best expose the banded structure of the input matrix. The result of the method is the original matrix M, on which we apply the permutation \(\pi \). However, the biMBA algorithm is non-optimal, heuristic, and does not find any permutation of columns (Garriga et al. 2011). To find both a permutation of columns and rows, the alternating biMBA method transposes the resulting matrix and iteratively repeats the described method on the transposed matrix until either convergence or reaching a predetermined number of steps. The alternating biMBA method clearly finds both a permutation of rows and a permutation of columns, however it is still (like the biMBA method) non-optimal and heuristic in nature. Also, this second iterative step comes with some price for some of the data described in this article: in the first data set, where neighboring columns of a matrix represent chromosome bands that are in physical proximity to one another, the goal may be to only find the optimal row permutation while not permuting the matrix columns.
As motivated by Fig. 2, finding a banded structure of a matrix will expose the cluster structure of the underlying data. The image of the banded structure can then be overlaid with a visualization of clusters, as described in Sect. 3.2. Because the rows of the matrix represent instances, highlighting one set of instances (one cluster) means highlighting several matrix rows. If the discovered clusters are exposed by the matrix structure, we can expect that several adjacent matrix rows will be highlighted, forming a wide band. Highlighting of clusters need not be limited to only one cluster: because each instance belongs to exactly one cluster, we can highlight them all at once. The only limitation is the number of clusters: because each cluster is colored with its own color, too many clusters may mean that colors will be too similar to each other to be distinguishable by the human eye.

The image of the clusters can also be overlaid with a visualization of the patterns explaining the clusters, presented in Sect. 3.3. If a chromosome band is discovered as an important chromosome band for the characterization of a cluster, we highlight the corresponding column. In the case of composite rules of the type

, both bands are understood as equally important and are therefore both highlighted. If a chromosome band appears in more than one rule, this is visualized by a stronger highlight of the corresponding matrix column. In the case of the ideal example, shown in Fig. 2, the second cluster is completely defined by having ones in columns 3, 4, 5, 6, and 7. We show this by highlighting these columns in the banded matrix. It is to be noted that the banded matrix visualization helps to determine if the clustering results are plausible. It also helps to identify the similarities and differences between clusters with respect to the patterns in the data.
4 Experimental data
In this section, we present the data sets which were used in the experiments. We first present a detailed explanation of multiresolution chromosomal amplification data, followed by the presentation of selected publicly available data sets that were previously used in Ristoski and Paulheim (2014).
4.1 Multiresolution chromosomal amplification data
A wide range of genetic mutations and molecular mechanisms known as chromosomal aberrations have been identified as the hallmarks of various disorders such as cancer, schizophrenia, and infertility (Albertson 2006; Vogelstein and Kinzler 2002). In cancer research, identification and characterization of chromosomal aberrations are crucial to study and understand pathogenesis of cancer. Furthermore, study of chromosomal aberrations provides necessary information to select the optimal target for cancer therapy on an individual level (Kirsch 1993). Study of chromosomal aberrations also has several clinical applications such as studying multiple congenital abnormalities and identifying the family history of Down syndrome (Obe and Vijayalaxmi 2007).
The data set we examined consists of DNA copy number amplifications in 4590 cancer patients. The data describes 4590 patients as data instances, with attributes being chromosomal locations indicating amplifications in the genome. These aberrations are described as 1’s (amplification) and 0’s (no amplification). Authors in Myllykangas et al. (2006) describe the amplification data set in detail. Amplification data is further described at two different resolution levels (312 and 393 locations, for 24 different chromosomes).
Given the complexity of the multiresolution data, we were forced to reduce the complexity of the learning setting to a simpler one, allowing us to develop and test the proposed methodology. To this end, we have reduced the size of the data set: from the initial set of instances describing 4590 patients, each belonging to one of the 73 different cancer types, we have focused on 34 most frequent cancer types only, as there were small numbers of instances available for many of the rare cancer types. This reduced the data set from 4590 instances to a 4104 instances. The choice of 34 most frequent cancers is motivated by the fact that it covers 90 % of the entire data set. Since the original data with 393 genomic locations are high dimensional and the results could be greatly affected by the curse of dimensionality (Bellman 1961), we partitioned the data into 24 different chromosomes and process each chromosome at a time. Additionally, chromosome-wise processing may help us find chromosome specific patterns for different cancer types. Nevertheless, this division is based on the assumption that the effects of amplifications on different chromosomes, produced by a cancer type, are independent. Similar to the experiments in Hollmén et al. (2003), which showed differences in frequent itemsets computed from one cluster at a time to the whole data set at once, we can expect different patterns when they are computed from one chromosome at a time to the whole data set at once.
In addition, in the experiments we have focused on a single chromosome (chromosome 1), using as input to step 2 of the proposed methodology the data clusters obtained at coarse resolution using a mixture modeling approach (Myllykangas et al. 2008).
When chromosomes are extracted from the data, some cancer patients show no amplifications in any regions of the chromosome 1. We have removed such samples without amplifications (zero vectors) because we are interested in the amplifications and their relation to cancers, not their absence. Considering negation cases is unsuitable because we are only investigating one chromosome at a time. A negation result could infer that if a region is not aberrated, it is likely to be a specific cancer which will be misleading as information from other chromosomes are missing. This reduces the sample size, for example sample size of chromosome 1 is reduced from 4104 to 407. While this data reduction may be an over-simplification, finding relevant patterns in this data set is a huge challenge, given the fact that even individual cancer types are known to consist of cancer sub-types which have not yet been explained in the medical literature. If we consider the entire data, inferencing and density estimation will produce degenerate results because of the curse of dimensionality (Bellman 1961). Additionally, the experiments performed on chromosome 1 can be seamlessly extended to all the other chromosomes, thus efficiently using each and every sample present in the data. Furthermore, chromosomewise analysis can generate chromosome specific patterns for certain cancer types. The proposed methodology may prove, in future work, to become a cornerstone in developing means through which such sub-types could be discovered, using automated pattern construction and innovative pattern visualization using banded matrices visualization.
In addition to the DNA amplifications data sets, we used supplementary background knowledge in the form of an ontology to enhance the analysis of the data set. The supplementary background knowledge consists of hierarchical structure of multiresolution amplification data, chromosomal locations of fragile sites, virus integration sites, cancer genes, and amplification hotspots. The hierarchical structure of multiresolution data is due to International System of Cytogenetic Nomenclature (ISCN) which allows the exact description of all numeric and structural amplifications in genomes (Shaffer and Tommerup 2005). A fragile site is a chromosomal region that tends to show a constriction or a gap and may tend to break on metaphase chromosomes when subjected to partial replication stress, i.e. following partial inhibition of DNA synthesis (Durkin and Glover 2007). A metaphase chromosome is a chromosome in the stage of the cell cycle (the sequence of events in the life of a cell) when a chromosome is most condensed, highly coiled, and aligned in the equator of the cell before being separated into each of the two daughter cells. At this stage chromosome is easiest to distinguish and study. Virus integration sites are also the chromosomal locations where viral DNA inserts into host-cell DNA (Hausen 2009). Approximately, 12 % of cancers are caused by viruses (Hausen 2009). Cancer genes are also the chromosome locations of known cancer causing genes. The list was obtained from Futreal et al. (2004). Amplification hotspots are frequently amplified chromosomal loci identified using computational modeling (Myllykangas et al. 2006).
4.2 Publicly available data sets
In addition to the chromosomal amplifications data, we tested our methodology on four publicly available data sets originally used in Ristoski and Paulheim (2014).
-
Cities Data set describes the most and least liveable cities in the world according the Mercer ranking.
-
NY Daily Data set describes the crawled news items along with their sentiment scores.
-
Tweets Data set is a collection of tweets with different features where the original task is to identify different sports related tweets.
-
Stumble Upon Data set consists of training data set used in the Kaggle competition.
To generate the hierarchical features, ‘DBpedia Direct Types’ ontology was used in the first three experiments, and the ‘Open directory project’ ontology was used to extract categories for each URL in the fourth data set, i.e. we used the same approach as in the original experiments reported in Ristoski and Paulheim (2014).
Since the data sets were highly sparse, we preprocessed the data to remove highly sparse variables. In the Cities data sets, we selected only those features which were positive in at least 25 different samples, but also eliminated features that were very dense, i.e. those that were positive in more than 170 instances. In the NY Daily data sets, we selected only the features that were positive in more than 200 samples but less than 450 samples. In the Tweets data set we selected only the features that were positive in more than 100 samples of the Tweets data set. Finally, in the Stumble Upon data set we selected only the features that were positive in more than 400 samples. Such preprocessing was motivated by the fact that features that are either very sparse or too dense carry very little information for class discrimination. Moreover, by removing these features we also mitigate the negative curse of dimensionality effects (Bellman 1961).
5 Experiments on multiresolution chromosomal amplification data
The following sections describe the results of running the developed three-step methodology on the chromosomal amplification data. We present the experimental results, result visualization and interpretation.
5.1 Mixture modeling
Mixture modeling itself consists of three steps: first we need to use model selection to determine the number of components (i.e. clusters) in the mixture model. Second, we need to learn the parameters of each component distributions, and finally, use the selected model to generate the data clusters. For the chromosomal amplification data set, we used the mixture models trained in our earlier contribution (Myllykangas et al. 2008). Through a model selection procedure documented in Tikka et al. (2007), the number of components for modeling chromosome 1 was set to \(J=6\).

Mixture model for chromosome 1. Only first 10 dimensions are shown for clarity. The figure just depicts a collection of numbers in the mixture model, which does not provide much insight to the expert
Figure 3 shows a visual illustration of the mixture model parameters for chromosome 1. In the figure, the first line denotes the number of components (J) in the mixture model and the data dimensionality (d). The lines beginning with # are comments and can be ignored. The fourth line shows the parameters of component distributions \((\pi _j)\) which are six probability values summing to 1. Similarly, the last six lines of the figure denote the parameters of the component distributions (\(\theta _{ji}\)). Figure 3 does not provide any insight into the data as it consists of many numbers and probability values. Therefore, we use banded matrix for visualization to demonstrate and evaluate the results produced by the mixture models and provide additional insights into the data set.
We clustered the data using the mixture model depicted in Fig. 3. Whereas the mixture model defines a probability model for the generation of data and can thus be used in soft clustering, allocating data vectors to the component densities that maximize the probability of data defines a hard clustering. Here, we focus on hard clustering of the samples of chromosomal amplification data, dividing the data set into six different clusters. The number of samples in each cluster are the following: \(|\text {Cluster }1|=30\), \(|\text {Cluster }2|=96|\), \(|\text {Cluster }3|=88\), \(|\text {Cluster }4|=81\), \(|\text {Cluster }5|=75\), \(|\text {Cluster }6|=37\).
5.2 Cluster visualization using banded matrices
We used the bidirectional minimal banded augmentation method, described in Sect. 3.4, to extract the banded structure in the data. As explained in Sect. 3.4, we decided to only allow permutations of rows of the data matrix. In Fig. 4, the black color indicates ones in the data and white color denotes zeros in the data. The resulting figure is then overlaid with the 6 clusters, discovered in Sect. 5.1.
By exposing the banded structure of a matrix, Fig. 4 allows a clear visualization of the clusters discovered in the data. Examination of Figs. 3 and 4 show that each cluster captures amplifications in some specific regions of the genome. Both figures capture a phenomenon that the p-arm of chromosome 1 (left part of the figure) shows a comparatively smaller number of amplifications whereas the q-arm shows a higher number of amplifications.
In Fig. 4, cluster 1 (component 1, \(\pi _{1}\)) is characterized by pronounced amplifications in the end of the q-arm (regions 1q32–q44) of chromosome 1. The figure also shows that samples in the second cluster (component 2, \(\pi _{2}\)) contain sporadic amplifications spread across both p and q-arms in different regions of chromosome 1. This cluster does not carry much information and contains cancer samples that do not show discriminating amplifications in chromosomes as the values of random variables are near 0.5. It is the only cluster that was split into many separate matrix regions. In contrast, cluster 3 (component 3, \(\pi _{3}\)) portrays marked amplifications in regions 1q11–44. Cluster 4 (component 4, \(\pi _{4}\)) shows amplifications in regions 1q21–25. Similarly, cluster 5 is denoted by amplifications in 1q21–25. The visualization with banded matrices in Fig. 4 also draws a distinction between clusters number 4 and 5, which upon first viewing show no obvious difference to the human eye. Cluster 6 (component 6, \(\pi _{6}\)) is defined by pronounced amplifications in the p-arm of chromosome 1.

Banded structure of the chromosome 1 data matrix with cluster information overlay
5.3 Rules induced through semantic pattern mining
Using the method described in Sect. 3.3, we induced subgroup descriptions for each cluster as the target class. For a selected cluster, all the other clusters represent the negative training examples, which resembles one-versus-all approach in multiclass classification. In this section, we discuss the results pertaining to clusters 1 and 3 (see Tables 1 and 2), while the rules for the other clusters, along with their visualization, are discussed in the following section. In our experiments we have considered only rules without negations in the rule conditions, as we are interested in the existence of amplifications characterizing the clusters and thereby the specific cancers (note that the absence of amplifications would mainly characterize the absence of cancers not their presence).
Tables 1 and 2 show the rules induced for clusters 1 and 3, together with their relevant statistics. The rules presented in Table 2 quantify the clustering results obtained in Sect. 5.1 and confirmed by banded matrix visualization in Sect. 5.2. The mixture model depicted in Fig. 3 and banded matrix visualization depicted in Figure 4 show that cluster 3 is marked by the amplifications in the regions 1q11–44. However, the rules obtained in Table 2 show that amplifications in all the regions 1q11–44 do not equally discriminate cluster 3. For example, rule

characterizes cluster 3 best with a precision of 1. This means that amplifications in regions 1q43–44 and 1q12 denote cluster 3. It also covers 81 of the 88 samples in cluster 3. Clinically, the amplifications in these regions characterises Ependymoma (Myllykangas et al. 2008).
Nevertheless, amplifications in regions 1q11–44 shown in Fig. 3 as discriminating regions, appear in at least one of the rules in Table 2 with varying degree of precision. The first part of the rule (i.e. amplifications in region 1q43–44) is the most discriminating for cluster 1 as shown in Table 1. However, with considerably reduced precision and lift.
Although the rule:

appears in semantic descriptions of both the clusters 1 and 3, addition of a conjunct 1q12 in the rule improves the discriminating power for cluster 3. Rule 2 covers all 88 samples of cluster 3 with precision of 0.77 whereas it covers 26 out of 30 samples in cluster 1 with the precision of 0.23. This shows that amplifications in region 1q43–44 characterize both clusters 1 and 3. If the negation rules are considered, amplifications only in regions 1q43–44 would more likely make it a candidate for cluster 1. Similarly, the second most discriminating rule for cluster 3 is:

which covers 78 positive samples and 9 negative samples.
The rules listed in Table 2 also capture the multiresolution phenomenon in the data. We input only one resolution of data to the algorithm but the hierarchy of different resolutions is made available to the algorithm as background knowledge. For example, the literal 1q43–44 denotes a joint region in coarse resolution thus showing that the algorithm produces results at different resolutions. The results at different resolutions improve the understandability and interpretability of the rules (Hollmén and Tikka 2007).
Furthermore, other information added to the background knowledge are amplification hotspots, fragile sites, cancer genes, which are discriminating features of cancers but do not show to discriminate any specific clusters present in the data. Therefore, such additional information can be better utilized in situations where the data set contains not only cancer samples but also control samples which is unfortunately not the situation here as our data set has only cancer patients.

Clusters 1 (left) and 3 (right) of the chromosome 1 data set with relevant columns highlighted. A highlighted column denotes that an amplification in the corresponding region characterizes the instances of the particular cluster. A darker hue means that the region appears in more rules. The numbers on top right of the figures correspond to rule numbers. For example, 1, 3 above rightmost column of cluster 3 indicates that the chromosome region appears in rules 1 and 3 tabulated in Table 2

Clusters 2 (left) and 4 (right) of the chromosome 1 data set with relevant columns highlighted
5.4 Visualizing semantic rules and clusters with banded matrices
The second way we can use the exposed banded structure of the data is to display columns that were found to be important due to appearing in rules from Sect. 5.3. We achieve this by highlighting the chromosomal regions which appear in the rules. Figure 5 depicts colored overlays of the rules on the ordered/serialized patient-chromosome matrix. As shown in Fig. 5, the highlighted band for cluster 1 spans chromosome regions 1q32–44. For cluster 3, the entire q-arm of the chromosome is highlighted, as indeed the instances in cluster 3 have amplifications throughout the entire arm. We can see that the regions 1q11–12 and 1q43–44 appear in rules with higher lift, in contrast to the other regions. This tells us that the amplifications on the edges of the region are more important for the characterization of the cluster (Table 2).
As shown in the left panel of Fig. 6, cluster 2 captures the heterogeneity in data (Table 3). Since, we are using only chromosome 1, this cluster is more likely to capture those cancers that are characterized by amplifications in chromosomes other than chromosome 1. The samples from clusters are distributed in different parts by the banded matrix visualization. The amplifications captured by this cluster are miscellaneous samples, i.e. those cancers that do not show prominent amplifications in chromosome 1. Nevertheless, amplifications captured by this cluster characterize glioblastoma multiforme (Myllykangas et al. 2008).
As shown in the right panel of Fig. 6, cluster 4 captures the amplifications near the beginning of the q arm of chromosome 1. The rules tabulated in Table 4 show amplifications in regions 1q21–1q25. Clinically, the amplifications in these regions of cluster 4 mark liposarcoma (Myllykangas et al. 2008) (Table 4).
The regions and rules in Cluster 5, depicted in the left panel of Fig. 7 overlap with the rules describing clusters 4. However, the rules describing these clusters have higher precision than those describing clusters 4 (Table 5). These two clusters are the prime candidates if any two clusters need to be merged. In terms of clinical relevance, the amplifications the regions captured by this cluster denotes malignant fibrous histiocytoma of bone (Myllykangas et al. 2008).

Clusters 5 (left) and 6 (right) of the chromosome 1 data set with relevant columns highlighted
The amplifications in the p-arm of Chromosome 1 captured by cluster 6 are depicted in the right panel of Fig. 7. This is clearly distinguishable from other clusters because other clusters mainly capture the amplifications in q-arm of chromosome 1. The amplification in these regions characterizes the phenomenon of small cell lung cancer (Myllykangas et al. 2008).
In summary, Figs. 4 and 5 together offer much more informative view of the structure of the underlying data than simply the list of rules in Table 6 or the cluster visualization in Fig. 3. The figure shows that most the samples in the same cluster also appear together in the banded matrix visualization even when we only allow permutations of rows in the data set. The figure, achieved by reordering the matrix rows by placing similar items closer together to form a banded structure, allows an easier visualization of the clusters and rules. It is important to reorder the rows independently of the clustering process. This is because the reordering does not depend on the cluster structure discovered. Therefore, the resulting figures offer new insight into both the data and the clustering.
6 Experiments on publicly available data sets
We repeated the experiments, using the developed pipeline on the publicly available data sets. In this section, we present the experimental results, their visualizations and interpretations for the four publicly available data sets.
6.1 Mixture modeling
Similar to the chromosome amplification data, we repeated the three steps (determining the number of clusters, learning the parameters of each component distribution and using the selected model to generate the clusters) for each of the publicly available data sets.
Following our previous work in Myllykangas et al. (2008), we used ten-fold cross-validation with cross-validated likelihood as the criteria for selection of the optimal number of clusters, similar to Tikka et al. (2007). In each data set, we trained mixture models in a cross-validation setting for the number of components ranging from 2 to 20 (30 and 50 in larger data sets NY Daily and Stumble Upon), with the assumption that there are at least two clusters in the data. Similarly, another assumption is that components greater than 20 (30 and 50 in NY Daily and Stumble Upon data) would overfit the data. Mixture models are susceptible to local optima, therefore, we train multiple models with the same number of components (50 in our experiments).
Figure 8 shows that for small numbers of clusters, the likelihood of mixture models increases smoothly until reaching a noticeable peak. For ideal data sets (seen in Tikka et al. (2007)), the peak represents a global maximum. Our experiments on real-world data sets show that identifying structures within data sets is not straight-forward. However, taking parsimony into account, even if larger numbers of components produce higher validataion likelihoods, we would select mixture models with a smaller number of components as they are computationally easier to train both in terms of time and memory and are also easily interpretable by the domain experts (Hollmén and Tikka 2007).

Model selection using ten-fold cross-validation in NY Daily, Tweets, Stumble Upon, and Cities data sets. The figure depicts averaged log-likelihood for training and validation sets. The interquartile range (IQR) for 50 different training and validation runs in ten-fold cross-validation setting have also been plotted. The number of clusters is determined as the point at which training and validation likelihoods depict a peak
By determining the smallest number of components for which the likelihood as seen in Fig. 8 of mixture models reaches a local peak, we select 6, 7, 4, and 10 components in the Tweets, NY Daily, Cities, and Stumble Upon data sets, respectively. Like in the case of chromosomal amplification data, we used the mixture model parameters for each data set to cluster it. We focused on hard clustering of the samples, dividing the data set into the number of clusters, determined in the previous step.
6.2 Cluster visualization using banded matrices
On the publicly available data sets, we ran the alternating biMBA method to expose the banded structure of the matrices. The choice of alternating method was motivated by the fact that the ordering of the columns in the publicly available data sets was arbitrary. This is unlike the amplification data set which had fixed ordering of regions in the genome.

The results of the methodology for the Cities data set. Top left the banded structure of the Cities data matrix with cluster information overlay. Top right cluster 2 of the Cities data set with relevant columns highlighted. Bottom rules for cluster 2 of the Cities data set
Cities The biMBA algorithm converged after 7 iterations exposing the banded structure of the matrix. The banded structure in Fig. 9 clearly visualizes the four clusters found by the presented methodology. Clusters 2 and 3 are almost completely separated from clusters 1 and 4. The visualization also shows that cluster 1 and cluster 2 are both composed of two parts which are hard to distinguish. This phenomenon was also captured during model selection in the Cities data set because the increase in validation likelihood was minimal when the number of components was increased from 3 to 4. When we selected four components, a relatively homogeneous cluster is broken down into two.
Tweets The biMBA algorithm converged after 33 iterations for the Twitter data set with credible results. The visualization provided in Fig. 10 shows that clusters 1, 2 and 3 are clearly separable from the rest of the data set. Cluster 4, the largest of the clusters, is split into two large parts, both of which are fairly homogeneous. However, clusters 5, 6, and 7 are relatively small with the value mixture components equal to 0.07, 0.05, and 0.03. Hence, these clusters are not fully exposed in the visualization.
NY Daily The biMBA algorithm converged after 11 iterations for the NY Daily data set. As seen in Fig. 11, it clearly highlights clusters 1, 2 and 6 and shows that clusters 4 and 3 are more similar to each other. Interestingly, even though cluster 3 is split into several parts, it can still be seen that the annotations, drawn on the left side of the visualization, are more important for cluster 3 (meaning that splitting the two clusters was a good choice). As in cluster 2 of the amplification data sets, the algorithm also highlights cluster 5 which does not capture a specific pattern but patterns scattered across different columns in the data set (Fig. 11).
Stumble Upon The Stumble Upon data set was the only data set on which our methodology did not achieve credible results. The model selection procedure shows that both training and validation likelihood smoothly increase until the number of components is 20. Even after the number of components was greater than 20, even the validation likelihood did not decrease showing that there is no apparent structure in the data as depicted in the bottom right panel of Fig. 8. The figure does not show high variation in likelihood among different number of components and also within each component among different runs because the number of data samples are high to constrain the mixture model. Similarly, the biMBA algorithm converged much more slowly than in the other data sets, taking 521 iterations to reach the optimal banded structure. Visualizing the structure shows that the data is fractured into several small chunks. Some clusters, like 8 and 10, are separated from the rest, but the remaining clusters are sporadically scattered across all the rows (Fig. 12).

The results of the methodology for the Tweets data set. Top left the banded structure of the Tweets data matrix with cluster information overlay. Top right cluster 1 of the Tweets data set with relevant columns highlighted. Bottom rules for cluster 1 of the Tweets data set

The results of the methodology for the NY Daily data set. Top left the banded structure of the NY Daily data matrix with cluster information overlay. Top right cluster 1 of the NY Daily data set with relevant columns highlighted. Bottom rules for cluster 1 of the NY Daily data set
6.3 Rules induced through semantic pattern mining
We ran the same semantic subgroup discovery procedure (with the same parameters) on the publicly available data sets as on the amplification data set. Due to the large amount of experimental results, we chose to describe one cluster and the top five rules for that cluster for each data set (Figs. 9, 11, 10). For the Stumble Upon data set, we did not describe the discovered cluster with rules because both the clustering and the banded structure visualization performed poorly on the data set.
Cities In cities dataset, cluster 2 was chosen as an example of a very well characterized cluster (Fig. 9). We report the top five rules, all which have 100 % precision. The first rule actually perfectly describes the cluster, since it covers all examples from cluster 2. By investigating the rule conjuncts it follows that this cluster contains cities that are at the same time annotated as centers, municipalities and populated places. Furthermore, the cities data set comes with a label describing its livability: low, medium, and high (Ristoski and Paulheim 2014). Although clustering, rule extraction, and visualization were performed independent of the labels, the rules and clusters mostly describe cities with medium and high livability. In the table we omit the full concept URIs for visual clarity. Nevertheless, the exact semantics of each concept can be verified by visiting the corresponding DBpedia pages, e.g., full URI of Center is http://dbpedia.org/class/yago/Center108523483.
NY Daily For this data sets, we report the top five rules for cluster 1 (Fig. 11). Similar to the previous data sets, the found rules are of high precision and each covers a relatively large portion of all examples from this cluster (a total of 107 examples). Compared to the subgroup descriptions found for the other five clusters, this cluster contains mainly headlines annotated with the District and Region concepts, together with Agent and Organism concepts.

The weakly banded structure of the Stumble Upon data set with cluster information overlay. Both the number of clusters and lack of a highly visible banded structure suggest a lack of structure in the data set
Tweets For this data set we feature the top five rules for cluster 1 (Fig. 10). The rules found were of lower precision (76–80 %), which indicates that this cluster is harder to describe compared to the clusters mentioned in the previous two data sets. Nevertheless, the subgroup descriptions indicate that this cluster contains mainly tweets mentioning specific athletes (i.e. annotated with Person and Athlete concepts), and not, for example, teams or organizations, which do appear in rules for the other clusters (e.g., Organization concept). Furthermore, the tweets data set consists of associated class labels which denotes sports related and unrelated tweets (Ristoski and Paulheim 2014). Although, clustering, rule extraction, and visualization were performed independent of the label, this cluster mostly contains tweets related to sports.
6.4 Visualizing semantic rules and clusters with banded matrices
Similar to the chromosomal amplifications data sets, we also highlighted the relevant variables captured by the rules describing each cluster on the public data sets. We visualized the top 5 rules for the three publicly available data sets on which the rule discovery algorithm was run (the NY Daily, Cities and Tweets data sets).
Cities Cluster 2 in the data set was perfectly described by the rules, This cluster was chosen as an example of a very well characterized cluster (Fig. 9). The visualization shows a clear band of features, with the top instances annotated by features on the left side of the chart and the bottom instances annotated by features on the right. Cluster 2 in the middle is characterized by containing instances that are annotated by features on both sides of the band, as instances above it are not annotated by the rightmost features and instances above are not annotated by the leftmost features. The visualization shows that all five top rules cover features on both sides of the band.
NY Daily For this data sets, we report the top five rules for cluster 1 as shown in Fig. 11. The visualization clearly identifies the banded structure of the data, with three distinct vertical bands. The cluster is characterized as the cluster which contains instances, annotated by the features in the (unlike clusters 3 and 4) second and (unlike clusters 2 and 6) third band. The visualisation shows that all rules take this into account as all rules explain cluster 1 with at least one conjunct covering features on the second band and one conjunct in the third band.
Tweets For this data set we feature the top five rules for cluster 1 (Fig. 10). Despite the lower precision of rules, extracted by our methodology, the visualization still clearly shows the most important features for cluster 1. The banded structure visualization shows us two sets of features that are important to cluster 1. The first is the block of tweets, annotated with the annotations Athlete and Contestant. One of these two annotations features in all top four rules, found for this cluster. The second, larger block of features is used in all top five rules we present. Additionally, the visualization of all clusters can also tell us why the precision of rules, found for this data set, was lower: cluster 2 contains several instances which are annotated by all features that also annotate features in cluster 1.
7 Stability analysis of clustering results
The success of the presented three-part methodology depends upon the results of cluster analysis. Since mixture models and clustering are unsupervised, which might result in different clustering solutions in different runs of the algorithm (Von Luxburg 2010). Therefore, it is imperative that we evaluate the stability of the results produced by our mixture models. In our experiments, we use the Expectation Maximization (EM) algorithm to learn the maximum likelihood parameters of those mixture models. An important property of Expectation Maximization algorithm is that it is deterministic for a given data set and a given initialization (McLachlan and Krishnan 2008). In other words, given the same data sets and same initialization, EM algorithm always converges on the same final model. However, one of the drawbacks of Expectation Maximization algorithm is that it is susceptible to local optima (McLachlan and Peel 2000). Therefore, we train the mixture model from random initialization multiple times to get the final result. In model selection, we consider the mean of the results and the dispersion to select the optimal number of components. In preparing the final model to use it for clustering, we train 200 different models from random initialization and select the one that produces the best likelihood as the final model for clustering.

Stability analysis of clustering results using four external measures of clustering
We have empirically evaluated the stability of our clustering results. We initially trained 100 mixture models initialized at random to convergence on the same data and measure the clustering accuracy, i.e. how often two observations belong to the same cluster. We could assume this setting to be a classification where first clustering solution to be the known class labels and each subsequent clustering labels as the classification produced by the model (Von Luxburg 2010). Since, we compare 100 models to each other there will be 4950 comparisons in all. In addition to clustering accuracy, we have also calculated other external measures of clustering quality such as the Jaccard index, Rand index, and Mantel statistics to determine the similarity in different clustering results produced by differently trained mixture models.
Results in Fig. 13 show that for unsupervised methods such as mixture models, the results of clustering are very stable. Clustering accuracy of approximately 70 % is a very good result in a multiclass classification setting. For example, the cities data set has 4 clusters, so a random classifier would generate only 25 % accuracy. Jaccard index and Rand index of more than 50 % also show that results are considerably stable. We calculate the Mantel statistics on the clustering results. The distance input to Mantel statistics is calculated from clustering labels obtained from two different clusterings. If two samples are in the same cluster, distance between them is 0, 1 otherwise. The matrices are positively correlated, and the associated p values are 0.001.
8 Summary and conclusions
The main contribution of this work is a three-part methodology for data analysis, consisting of (i) data clustering with mixture models, (ii) extraction of semantic patterns (rules) from the clusters, using an ontology of relationships between the different resolutions of the multiresolution data, and (iii) integration of the results in a visual display, illustrating the clusters, and the identified rules by visualizing them over the banded matrix structure. The proposed visualization allows us to explain the discovered patterns by combining different views of the data, which may be difficult to compare without a unifying visual display. In our experiments, we analyzed DNA copy number amplifications in the form of 0–1 data, where the clustering developed in previous work was augmented by explanatory rules derived from a semantic pattern mining approach combined by the facility to display the bandedness structure of the data.
Our experiments with using the proposed algorithm on the NY Daily, Tweets and Cities data sets also demonstrate the wide usability of the algorithm which extends beyond the original application to DNA copy number amplifications onto any data set annotated by a hierarchically ordered set of background knowledge nodes. The results on the Stumble Upon data set, while at first glance a negative result, also give important insight into the data set. Because all three algorithms (clustering, rule search and banded visualization) performed equally badly on the data set, we can with a much higher confidence claim that no particular structure in exists in the data set.
The proposed semi-automated methodology provides complete analysis of a complex real-world multiresolution data. The results produced in the form of different clusters, rules, and visualizations with the help of banded matrices are made interpretable for the domain experts. Especially, the visualizations with banded matrix helps to understand the clustering results and the rules generated by the semantic pattern mining algorithm. Furthermore, the background knowledge used to supplement semantic data mining algorithm enables us to analyze multiresolution data and garner results at different levels of multiresolution hierarchy. Similarly, the rules obtained by semantic data mining algorithm helps to quantitatively prioritize chromosomal regions that are hallmarks of certain cancers among different chromosomal regions that are amplified in those cancer patients.
The proposed approach accepts as input single-resolution data but allows for multiresolution data analysis due to the hierarchy of regions used as background knowledge in semantic pattern mining algorithm. In the future, we plan to develop a system to directly accept multiresolution data as input. Similarly, we will consider the cancer instance labels, since in the present work we focused only on cluster labels. In future work, we plan to formulate the problem as a multiclass classification problem in the semantic pattern mining setting as learning from ambiguous labels or partial labels and in mixture model clustering setting as soft clustering problem. Furthermore, another direction of research is to reformulate the banded matrix problem to consider class labels and directly benefit from cancer or cluster labels.
Similarly, we could also reformulate the instance descriptions by adding the truth values of the pattern alongside the original attributes and then compute the mixture model. Furthermore, the methodology is evaluated on data sets (different data sets denoting different chromosomes) on a single application area, i.e. chromosomal amplifications in cancer genomics.
References
Adhikari, P. R., & Hollmén, J. (2010). Patterns from multiresolution 0–1 data. In Proceedings of the ACM SIGKDD workshop on useful patterns (UP ’10) (pp. 8–16). New York: NY, USA, ACM.
Adhikari, P. R., & Hollmén, J. (2013). Mixture models from multiresolution 0–1 data. In J. Fürnkranz, E. Hüllermeier, & T. Higuchi (Eds.), Proceedings of Sixteenth International Conference on Discovery Science (DS 2013), Lecture Notes in Computer Science (Vol. 8140, pp. 1–16). Berlin, Heidelberg: Springer.
Adhikari, P. R., Vavpetič, A., Kralj, J., Lavrač, N., & Hollmén, J. (2014). Explaining mixture models through semantic pattern mining and banded matrix visualization. In S. Džeroski, P. Panov, D. Kocev, & L. Todorovski (Eds.), Discovery science, Lecture Notes in Computer Science (Vol. 8777, pp. 1–12). Springer International Publishing.
Agrawal, R., & Srikant, R. (1994, September 12–15). Fast algorithms for mining association rules in large databases. In J. B. Bocca, M. Jarke, & C. Zaniolo (Eds.), Proceedings of the 20th international conference on very large data bases (pp. 487–499). San Francisco, CA, USA. Morgan Kaufmann.
Albertson, D. G. (2006). Gene amplification in cancer. Trends in Genetics, 22(8), 447–455.
Bellman, R. E. (1961). Adaptive control processes—A guided tour. Princeton, NJ: Princeton University Press.
Bishop, C. M. (2006). Pattern recognition and machine learning (information science and statistics). Secaucus, NJ: Springer.
Celeux, G. (2007). Mixture models for classification. In R. Decker & H.-J. Lenz (Eds.), Advances in data analysis, studies in classification, data analysis, and knowledge organization (pp. 3–14). Berlin, Heidelberg: Springer.
Chen, C.-H., Hwu, H.-G., Jang, W.-J., Kao, C.-H., Tien, Y.-J., Tzeng, S. L., et al.(2004). Matrix visualization and information mining. In J. Antoch, (Eds.), Proceedings in computational statistics (COMPSTAT 2004) (pp. 85–100). Physica-Verlag HD.
Chen, J., & Khalili, A. (2008). Order selection in finite mixture models with a nonsmooth penalty. Journal of the American Statistical Association, 103(484), 1674–1683.
Cherkassky, V. S., & Mulier, F. (1998). Learning from data: Concepts, theory, and methods (1st ed.). New York, NY: Wiley.
Gene Ontology Consortium. (2008). The gene ontology project in 2008. Nucleic Acids Research, 36(Database-Issue), 440–444.
Dempster, A. P., Laird, N. M., & Rubin, D. B. (1977). Maximum likelihood from incomplete data via the EM algorithm. Journal of the Royal Statistical Society, Series B, 39(1), 1–38.
Despierre, E., Lambrechts, D., Neven, P., Amant, F., Lambrechts, S., & Vergote, I. (2010). The molecular genetic basis of ovarian cancer and its roadmap towards a better treatment. Gynecologic Oncology, 117(2), 358–365.
D’haene, B., Vandesompele, J., & Hellemans, J. (2010). Accurate and objective copy number profiling using real-time quantitative PCR. Methods, 50(4), 262–270.
Durkin, S. G., & Glover, T. W. (2007). Chromosome fragile sites. Annual Review of Genetics, 41(1), 169–192.
Figueiredo, M. A. T., & Jain, A. K. (2002). Unsupervised learning of finite mixture models. IEEE Transactions on Pattern Analysis and Machine Intelligence, 24(3), 381–396.
Fisher, R. A. (1922). On the interpretation of \(\chi ^{2}\) from contingency tables, and the calculation of p. Journal of the Royal Stat. Society, 85(1), 87–94.
Futreal, P. A., Coin, L., Marshall, M., Down, T., Hubbard, T., Wooster, R., et al. (2004). A census of human cancer genes. Nature Reviews Cancer, 4(3), 177–183.
Garriga, G. C., Junttila, E., & Mannila, H. (2011). Banded structure in binary matrices. Knowledge and Information Systems, 28(1), 197–226.
Hamming, R. W. (1986). Numerical methods for scientists and engineers (2nd ed.). New York, NY: Dover.
Hand, D., Mannila, H., & Smyth, P. (2001). Principles of data mining. Adaptive computation and machine learning series. Cambridge: MIT Press.
Hastie, T., Tibshirani, R., & Friedman, J. (2009). The elements of statistical learning: Data mining, inference, and prediction (2nd ed.). New York: Springer.
Hausen, H. Z. (2009). The search for infectious causes of human cancers: Where and why. Virology, 392(1), 1–10.
Hämäläinen, W. (2010). Efficient search for statistically significant dependency rules in binary data. PhD thesis, Department of Computer Science, University of Helsinki, Finland.
Hollmén, J., Seppänen, J. K., & Mannila, H. (2003) Mixture models and frequent sets: Combining global and local methods for 0–1 data. In Proceedings of the third SIAM international conference on data mining (pp. 289–293). Society of Industrial and Applied Mathematics.
Hollmén, J., & Tikka, J. (2007). Compact and understandable descriptions of mixture of Bernoulli distributions. In M. R. Berthold, J. Shawe-Taylor, & N. Lavrač (Eds.), Proceedings of the 7th international symposium on intelligent data analysis (IDA 2007), Lecture Notes in Computer Science (Vol. 4723, pp. 1–12). Ljubljana, Slovenia: Springer.
Holm, S. (1979). A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics, 6(2), 65–70.
Hotho, A., Staab, S., & Stumme, G. (2003). Explaining text clustering results using semantic structures. In N. Lavrač, D. Gamberger, L. Todorovski, & H. Blockeel (Eds.), Knowledge Discovery in Databases: 7th European Conference on Principles and Practice of Knowledge Discovery in Databases (PKDD 2003), LNAI (Vol. 2838, pp. 217–228). Springer.
Huang, T., Peng, H., & Zhang, K. (2013). Model Selection for Gaussian mixture models. arXiv preprint arXiv:1301.3558.
Jain, A. K., Murty, M. N., & Flynn, P. J. (1999). Data clustering: a review. ACM Computing Surveys (CSUR), 31(3), 264–323.
Jawerth, B., & Sweldens, W. (1994). An overview of wavelet based multiresolution analyses. SIAM Review, 36(3), 377–412.
Kirsch, I. R. (1993). The causes and consequences of chromosomal aberrations. Boca Raton: CRC Press.
Klösgen, W. (1996). Explora: A multipattern and multistrategy discovery assistant. In U. M. Fayyad, G. Piatetsky-Shapiro, P. Smyth, & R. Uthurusamy, (Eds.), Advances in knowledge discovery and data mining (pp. 249–271). American Association for Artificial Intelligence.
Knuutila, S., Aalto, Y., Autio, K., Björkqvist, A., El-Rifai, W., Hemmer, S., et al. (1999). DNA copy number losses in human neoplasms. Gynecologic Oncology, 155(2), 683–694.
Kononenko, I., & Kukar, M. (2007). Machine learning and data mining: Introduction to principles and algorithms. West Sussex: Horwood Publishing Limited.
Kuhn, M., & Johnson, K. (2013). Applied Predictive Modeling. New York, Heidelberg, Dordrecht, London: Springer.
Langohr, L., Podpečan, V., Petek, M., Mozetič, I., Gruden, K., Lavrač, N., et al. (2013). Contrasting subgroup discovery. The Computer Journal, 56(3), 289–303.
Lau, J. H., Grieser, K., Newman, D., & Baldwin, T. (2011). Automatic labelling of topic models. In Proceedings of the 49th annual meeting of the association for computational linguistics: Human language technologies (Vol. 1, pp. 1536–1545). Association for Computational Linguistics.
Lavrač, N., Kavšek, B., Flach, P. A., & Todorovski, L. (2004). Subgroup discovery with CN2-SD. Journal of Machine Learning Research, 5, 153–188.
Lawrynowicz, A., & Potoniec, J. (2011). Fr-ONT: An algorithm for frequent concept mining with formal ontologies. In M. Kryszkiewicz, H. Rybinski, A. Skowron, & Z. W. Raś (Eds.), Foundations of intelligent systems. Proceedings of 19th international symposium on methodologies for intelligent systems (ISMIS 2011), Lecture Notes in Computer Science (Vol. 6804, pp. 428–437). Berlin, Heidelberg: Springer.
Lindeberg, T. (1994). Scale-space theory: A basic tool for analysing structures at different scales. Journal of Applied Statistics, 21(2), 224–270.
Liu, B., Hsu, W., & Ma, Y. (1998, August) Integrating classification and association rule mining. In Proceedings of the 4th international conference on knowledge discovery and data mining (KDD’98) (pp. 80–86). AAAI Press.
Maglott, D., Ostell, J., Pruitt, K. D., & Tatusova, T. (2005). Entrez Gene: gene-centered information at NCBI. Nucleic Acids Research, 33, D54–D58.
McLachlan, G. J. (1987). On bootstrapping the likelihood ratio test stastistic for the number of components in a normal mixture. Journal of the Royal Statistical Society, Series C (Applied Statistics), 36(3), 318–324.
McLachlan, G. J., & Krishnan, T. (2008). The EM algorithm and extensions. Wiley series in probability and statistics (2nd ed.). New York: Wiley.
McLachlan, G. J., & Peel, D. (2000). Finite mixture models, probability and statistics–Applied probability and statistics section (Vol. 299). New York: Wiley.
Mei, Q., Shen, X., & Zhai, C. X. (2007). Automatic labeling of multinomial topic models. In Proceedings of the 13th ACM SIGKDD international conference on knowledge discovery and data mining (pp. 490–499). ACM.
Melnykov, V., & Maitra, R. (2010). Finite mixture models and model-based clustering. Statistics Surveys, 4, 80–116.
Mukherjee, D., Wu, Q. M. J., & Nguyen, T. M. (2013). Multiresolution based Gaussian mixture model for background suppression. IEEE Transactions on Image Processing, 22(12), 5022–5035.
Myllykangas, S., Himberg, J., Böhling, T., Nagy, B., Hollmén, J., & Knuutila, S. (2006). DNA copy number amplification profiling of human neoplasms. Oncogene, 25(55), 7324–7332.
Myllykangas, S., Tikka, J., Böhling, T., Knuutila, S., & Hollmén, J. (2008). Classification of human cancers based on DNA copy number amplification modeling. BMC Medical Genomics, 1(15), 1.
Novak, P., Lavrač, N., & Webb, G. I. (2009). Supervised descriptive rule discovery: A unifying survey of contrast set, emerging pattern and subgroup mining. Journal of Machine Learning Research, 10, 377–403.
Obe, G., & Vijayalaxmi, (2007). Chromosomal alterations: Methods, results, and importance in human health. Berlin: Springer.
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., & Kanehisa, M. (1999). KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Research, 27(1), 29–34.
Oliveira-Brochado, A., & Martins, F. V. (2005). Assessing the number of components in mixture models: A review. FEP Working papers 194, Universidade do Porto, Faculdade de Economia do Porto.
Panov, P. (2012, July) A modular ontology of data mining. Doctoral dissertation, Jožef Stefan International Postgraduate School.
Piatetsky-Shapiro, G. (1991). Discovery, analysis, and presentation of strong rules. In G. Piatetsky-Shapiro & W. J. Frawley (Eds.), Knowledge discovery in databases. Cambridge: AAAI/MIT Press.
Pollack, J. R., Perou, C. M., Alizadeh, A. A., Eisen, M. B., Pergamenschikov, A., Williams, C. F., et al. (1999). Genome-wide analysis of DNA copy-number changes using cDNA microarrays. Nature Genetics, 23(1), 41–46.
Rancoita, P. M. V., Hutter, M., Bertoni, F., & Kwee, I. (2009). Bayesian DNA copy number analysis. BMC Bioinformatics, 10(1), 10.
Ristoski, P., & Paulheim, H. (2014). Feature selection in hierarchical feature spaces. In S. Džeroski, P. Panov, D. Kocev, & L. Todorovski, (Eds.), Discovery science, Lecture Notes in Computer Science (Vol. 8777, pp. 288–300). Springer International Publishing.
Shaffer, L. G., & Tommerup, N. (2005). ISCN 2005: An international system for human cytogenetic nomenclature (2005) recommendations of the international standing committee on human cytogenetic nomenclature. Basel: Karger.
Smyth, P. (2000). Model selection for probabilistic clustering using cross-validated likelihood. Statistics and Computing, 10(1), 63–72.
Srinivasan, A. (2007, March). Aleph Manual.
Sugiyama, K., Tagawa, S., & Toda, M. (1981). Methods for visual understanding of hierarchical system structures. IEEE Transactions on Systems, Man, and Cybernetics, 11(2), 109–125.
Thrun, S. (1995). Extracting rules from artificial neural networks with distributed representations. In G. Tesauro, D. S. Touretzky, & T. K. Leen (Eds.), Advances in neural information processing systems (Vol. 7, pp. 505–512). Cambridge: MIT Press.
Tikka, J., Hollmén, J., & Myllykangas, S. (2007). Mixture modeling of DNA copy number amplification patterns in cancer. In F. Sandoval, A. Prieto, J. Cabestany, & M. Graña (Eds.), Proceedings of the 9th international work-conference on artificial neural networks (IWANN 2007), Lecture Notes in Computer Science (Vol. 4507, pp. 972–979). San Sebastián, Spain: Springer.
Trajkovski, I., Lavrač, N., & Tolar, J. (2008). SEGS: Search for enriched gene sets in microarray data. Journal of Biomedical Informatics, 41(4), 588–601.
Trajkovski, I., Železný, F., Lavrač, N., & Tolar, J. (2008). Learning relational descriptions of differentially expressed gene groups. IEEE Transactions on Systems, Man, and Cybernetics, Part C, 38(1), 16–25.
Tresp, V., Hollatz, J., & Ahmad, S. (1997). Representing probabilistic rules with networks of gaussian basis functions. Machine Learning, 27(2), 173–200.
Tufte, E. R. (1986). The visual display of quantitative information. Cheshire, CT: Graphics Press.
Vavpetič, A., & Lavrač, N. (2013). Semantic subgroup discovery systems and workflows in the SDM-toolkit. The Computer Journal, 56(3), 304–320.
Vavpetič, A., Novak, P. K., Grčar, M., Mozetič, I., & Lavrač, N. (2013). Semantic data mining of financial news articles. In J. Fürnkranz, E. Hüllermeier, & T. Higuchi (Eds.), Proceedings of sixteenth international conference on discovery science (DS 2013), Lecture Notes in Computer Science (Vol. 8140, pp. 294–307). Berlin, Heidelberg: Springer.
Vavpetič, A., Podpečan, V., & Lavrač, N. (2014). Semantic subgroup explanations. Journal of Intelligent Information Systems, 42(2), 233–254.
Vogelstein, B., & Kinzler, K. W. (2002). The genetic basis of human cancer. New York: McGraw-Hill.
Von Luxburg, U. (2010). Clustering stability: An overview. Foundations and Trends in Machine Learning, 2(3), 235–274.
Weinan, E. (2011). Principles of multiscale modeling. Cambridge: Cambridge University Press.
Willsky, A. S. (2002). Multiresolution markov models for signal and image processing. Proceedings of the IEEE, 90(8), 1396–1458.
Wilson, R. (2000) MGMM: Multiresolution Gaussian mixture models for computer vision. In Proceedings of 15th international conference on pattern recognition (Vol. 1, pp. 212–215).
Woo, M.-J., & Sriram, T. N. (2006). Robust estimation of mixture complexity. Journal of the American Statistical Association, 101(476), 1475–1486.
Wrobel, S. (1997). An algorithm for multi-relational discovery of subgroups. In Proceedings of the first European conference on principles of data mining and knowledge discovery (PKDD ’97), (pp. 78–87). Springer.
Wu, H.-M., Tien, Y.-J., & Chen, C.-H. (2010). GAP: A graphical environment for matrix visualization and cluster analysis. Computational Statistics and Data Analysis, 54(3), 767–778.
Acknowledgments
This work was supported by Helsinki Doctoral Programme in Computer Science—Advanced Computing and Intelligent Systems (Hecse) and by the Slovenian Ministry of Higher Education, Science and Technology grants. Additionally, the work was supported by the Academy of Finland (Grant Number 258568) and partially supported by the European Commission through the Human Brain Project (Grant Number 604102). Thanks to Petar Ristoski for providing us with his data set exports.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editors: Saso Dzeroski, Dragi Kocev, and Pance Panov.
Rights and permissions
About this article

Cite this article
Adhikari, P.R., Vavpetič, A., Kralj, J. et al. Explaining mixture models through semantic pattern mining and banded matrix visualization. Mach Learn 105, 3–39 (2016). https://doi.org/10.1007/s10994-016-5550-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10994-016-5550-3