
Abstract
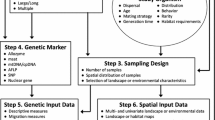
Landscape genetics is an emerging interdisciplinary field that combines methods and concepts from population genetics, landscape ecology, and spatial statistics. The interest in landscape genetics is steadily increasing, and the field is evolving rapidly. We here outline four major challenges for future landscape genetic research that were identified during an international landscape genetics workshop. These challenges include (1) the identification of appropriate spatial and temporal scales; (2) current analytical limitations; (3) the expansion of the current focus in landscape genetics; and (4) interdisciplinary communication and education. Addressing these research challenges will greatly improve landscape genetic applications, and positively contribute to the future growth of this promising field.
Similar content being viewed by others

References
Balkenhol N, Waits LP, Dezzani R (2009) Statistical approaches in landscape genetics: an evaluation of methods for linking landscape and genetic data. Ecography. doi:10.1111/j.1600-0587.2009.05807.x (in press)
Bolnick DI, Fitzpatrick BM (2007) Sympatric speciation: models and empirical evidence. Annu Rev Ecol Evol Syst 38:459–487. doi:10.1146/annurev.ecolsys.38.091206.095804
Brumfield RT, Beerli P, Nickerson DA, Edwards SV (2003) The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol Evol 18:249–256. doi:10.1016/S0169-5347(03)00018-1
Chen C, Durand E, Forbes F, François O (2007) Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7:747–756. doi:10.1111/j.1471-8286.2007.01769.x
Coulon A, Cosson JF, Angibault JM et al (2004) Landscape connectivity influences gene flow in a roe deer population inhabiting a fragmented landscape: an individual-based approach. Mol Ecol 13:2841–2850. doi:10.1111/j.1365-294X.2004.02253.x
Cushman SA (2006) Effects of habitat loss and fragmentation on amphibians: a review and prospectus. Biol Conserv 128:231–240. doi:10.1016/j.biocon.2005.09.031
Cushman SA, McKelvey KS, Hayden J, Schwartz MK (2006) Gene flow in complex landscapes: testing multiple hypotheses with causal modeling. Am Nat 168:486–499. doi:10.1086/506976
Cushman SA, McKelvey KS, Schwartz MK (2009) Using empirically derived source-destination models to map regional conservation corridors. Conserv Biol. doi:10.1111/j.1523-1739.2008.01111.x (in press)
Davies KF, Gascon C, Margules CR (2001) Habitat fragmentation: consequences, management, and future research priorities. In: Soule ME, Orians G (eds) Conservation biology: research priorities for the next decade. Island Press, Washington, DC, pp 81–98
Dupanloup I, Schneider S, Excoffier L (2002) A simulated annealing approach to define genetic structure of populations. Mol Ecol 11:2571–2581. doi:10.1046/j.1365-294X.2002.01650.x
Dyer RJ, Nason JD (2004) Population graphs: the graph-theoretic shape of genetic structure. Mol Ecol 13:1713–1728. doi:10.1111/j.1365-294X.2004.02177.x
Epps CW, Wehausen JD, Bleich VC et al (2007) Optimizing dispersal and corridor models using landscape genetics. J Appl Ecol 44:714–724. doi:10.1111/j.1365-2664.2007.01325.x
Faubet P, Gaggiotti OE (2008) A new Bayesian method to identify the environmental factors that influence recent migration. Genetics 178:1491–1504. doi:10.1534/genetics.107.082560
Foll M, Gaggiotti OE (2006) Identifying the environmental factors that determine the genetic structure of populations. Genetics 174:875–891. doi:10.1534/genetics.106.059451
François O, Ancelet S, Guillot G (2006) Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics 174:805–816. doi:10.1534/genetics.106.059923
Futuyma DJ (1997) Evolutionary biology. Sinauer, Sunderland
Geffen E, Anderson MJ, Wayne RK (2004) Climate and habitat barriers to dispersal in the highly mobile grey wolf. Mol Ecol 13:2481–2490. doi:10.1111/j.1365-294X.2004.02244.x
Holderegger R, Wagner HH (2006) A brief guide to landscape genetics. Landscape Ecol 21:793–796. doi:10.1007/s10980-005-6058-6
Holderegger R, Wagner HH (2008) Landscape genetics. Bioscience 58:199–207. doi:10.1641/B580306
Holderegger R, Kamm U, Gugerli F (2006) Adaptive versus neutral genetic diversity: implications for landscape genetics. Landscape Ecol 21:797–807. doi:10.1007/s10980-005-5245-9
Holderegger R, Hermann D, Poncet B et al (2008) Land ahead: using genome scans to identify molecular markers of adaptive relevance. Plant Ecol Div 1:273–283
Holzhauer S, Ekschmitt K, Sander A-C et al (2006) Effect of historic landscape change on the genetic structure of the bush-cricket Metrioptera roeseli. Landscape Ecol 21:891–899. doi:10.1007/s10980-005-0438-9
Joost S, Bonin A, Bruford MW et al (2007) A spatial analysis method (SAM) to detect candidate loci for selection: towards a landscape genomics approach to adaptation. Mol Ecol 16:3955–3969. doi:10.1111/j.1365-294X.2007.03442.x
Kamm U (2008) Landscape genetics of a rare, naturally scattered, temperate forest tree (Sorbus domestica). PhD-thesis, ETH Zürich, Zürich, Switzerland
Keyghobadi N, Roland J, Matter S, Strobeck C (2005) Among- and within-patch components of genetic diversity respond at different rates to habitat fragmentation: an empirical demonstration. Proc R Soc Ser B 272:553–560. doi:10.1098/rspb.2004.2976
Landergott U, Holderegger R, Kozlowski G, Schneller JJ (2001) Historical bottlenecks decrease genetic diversity in natural populations of Dryopteris cristata. Heredity 87:344–355. doi:10.1046/j.1365-2540.2001.00912.x
Latch E, Dharmarajan G, Glaubitz JC, Rhodes OE Jr (2006) Relative performance of Bayesian clustering software for inferring population substructure and individual assignment at low levels of population differentiation. Conserv Genet 7:295–302. doi:10.1007/s10592-005-9098-1
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. Trends Ecol Evol 18:189–197. doi:10.1016/S0169-5347(03)00008-9
Mayr E (1954) Change of genetic environment and evolution. In: Huxley J, Hardy AC, Ford EB (eds) Evolution as a process. Allen and Unwin, London, pp 157–180
Mayr E (1988) Processes of speciation in animals. In: Mayr E (ed) Towards a new philosophy of biology: observations of an evolutionist. Harvard University Press, Cambridge, pp 364–382
McRae BH (2006) Isolation by resistance. Evol Int J Org Evol 60:1551–1561
McRae BH, Beier P (2007) Circuit theory predicts gene flow in plant and animal populations. Proc Natl Acad Sci USA 104:19885–19890. doi:10.1073/pnas.0706568104
McRae BH, Dickson BG, Keitt TH (2008) Using circuit theory to model connectivity in ecology and conservation. Ecology 89:2712–2724. doi:10.1890/07-1861.1
Meudt HM, Clarke AC (2007) Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci 12:106–117. doi:10.1016/j.tplants.2007.02.001
Morin PA, Luikart G, Wayne RK et al (2004) SNPs in ecology, evolution and conservation. Trends Ecol Evol 19:208–216. doi:10.1016/j.tree.2004.01.009
Orsini L, Corander J, Alasentie A, Hanski I (2008) Genetic spatial structure in a butterfly metapopulation correlates better with past than present demographic structure. Mol Ecol 17:2629–2642. doi:10.1111/j.1365-294X.2008.03782.x
Ouborg NJ, Vriezen WH (2007) An ecologist’s guide to ecogenomics. J Ecol 95:8–16. doi:10.1111/j.1365-2745.2006.01197.x
Pascual-Hortal L, Saura S (2007) Impact of spatial scale on the identification of critical habitat patches for the maintenance of landscape connectivity. Landsc Urban Plan 83:176–186. doi:10.1016/j.landurbplan.2007.04.003
Petit R, Vendramin GG (2007) Plant phylogeography based on organelle genes: an introduction. In: Weiss S, Ferrand N (eds) Phylogeography of Southern European refugia—evolutionary perspectives on the origins and conservation of European biodiversity. Springer, Dordrecht, pp 23–97
Petit RJ, Duminil J, Fineschi S et al (2005) Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol Ecol 14:689–701. doi:10.1111/j.1365-294X.2004.02410.x
Pritchard JK, Stephens M, Peter D (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rico Y, Lorenzo C, González-Cózatl FX et al (2008) Phylogeography and population structure of the endangered Tehuantepec jackrabbit Lepus flavigularis: implications for conservation. Conserv Genet 9:1467–1477. doi:10.1007/s10592-007-9480-2
Rousset F (2000) Genetic differentiation between individuals. J Evol Biol 13:58–62. doi:10.1046/j.1420-9101.2000.00137.x
Scandura M, Iacolina L, Crestanello B et al (2008) Ancient versus recent processes as factors shaping the genetic variation of the European wild boar: are the effects of the last glaciation still detectable? Mol Ecol 17:1745–1762. doi:10.1111/j.1365-294X.2008.03703.x
Schlesinger WH, Clark JS, Mohan JE, Reid CD (2001) Global environmental change: effects on biodiversity. In: Soule ME, Orians G (eds) Conservation biology: research priorities for the next decade. Island Press, Washington, DC, pp 175–224
Schwartz MK, McKelvey KS (2009) Why sampling scheme matters: the effect of sampling scheme on landscape genetic results. Conserv Genet. doi:10.1007/s10592-008-9622-1
Selkoe KA, Toonen RJ (2006) Microsatellites for ecologists: a practical guide to using and evaluating microsatellite markers. Ecol Lett 9:615–629. doi:10.1111/j.1461-0248.2006.00889.x
Spear SF, Peterson CR, Matoq MD, Storfer A (2005) Landscape genetics of the blotched tiger salamander (Ambystoma tigrinum melanostictum). Mol Ecol 14:2553–2564. doi:10.1111/j.1365-294X.2005.02573.x
Stephens PA, Buskirk SW, del Rio CM (2007) Inference in ecology and evolution. Trends Ecol Evol 22:192–197. doi:10.1016/j.tree.2006.12.003
Storfer A, Murphy MA, Evans JS et al (2007) Putting the “landscape” in landscape genetics. Heredity 98:128–142. doi:10.1038/sj.hdy.6800917
Vandergast AG, Bohonak AJ, Weissman DB, Fisher RN (2007) Understanding the genetic effects of recent habitat fragmentation in the context of evolutionary history: phylogeography and landscape genetics of a southern California endemic jerusalem cricket (Orthoptera: Stenopelmatidae: Stenopelmatus). Mol Ecol 16:977–992. doi:10.1111/j.1365-294X.2006.03216.x
Vasemägi A, Primmer CR (2005) Challenges for identifying functionally important genetic variation: the promise of combining complementary research strategies. Mol Ecol 14:3623–3642. doi:10.1111/j.1365-294X.2005.02690.x
Via S (2001) Sympatric speciation in animals: the ugly duckling grows up. Trends Ecol Evol 16:381–390. doi:10.1016/S0169-5347(01)02188-7
Wagner H, Fortin M-J (2005) Spatial analysis of landscapes: concepts and statistics. Ecology 86:1975–1987. doi:10.1890/04-0914
Whitham TG, Bailey JK, Schweitzer JA et al (2006) A framework for community and ecosystem genetics: from genes to ecosystems. Nat Rev Genet 7:510–523. doi:10.1038/nrg1877
Wright S (1977) Evolution and the genetics of populations. University of Chicago Press, Chicago
Wu J (2007) Scale and scaling: a cross-disciplinary perspective. In: Wu J, Hobbs RJ (eds) Key topics in landscape ecology. Cambridge University Press, Cambridge, pp 115–142
Acknowledgments
We thank Brad McRae and two anonymous reviewers for valuable comments that helped to improve this manuscript. We also thank the organizers of the 2007 IALE World Congress.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Participants of the Landscape Genetics Research Agenda Workshop, held at the 2007 World Congress of the International Association of Landscape Ecologists (IALE), in Wageningen, The Netherlands: Paul Arens, Pascal Campagne, Virginia H. Dale, Alfredo G. Nicieza, Marinus J. M. Smulders, Edoardo Tedesco, Hongfang Wang, Tzeidle Wasserman.
Rights and permissions
About this article
Cite this article
Balkenhol, N., Gugerli, F., Cushman, S.A. et al. Identifying future research needs in landscape genetics: where to from here?. Landscape Ecol 24, 455–463 (2009). https://doi.org/10.1007/s10980-009-9334-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10980-009-9334-z