
Abstract
La2O3–SiO2 powders were prepared by the sol–gel and combustion method without addition of strong polar solvents to the initial system. Changes in the crystalline La2O3 structure and modification of surface binary oxides system were described as a function of concentration of the second component, that is silicon dioxide. A series of samples of La to Si molar ratios of 1:0.0, 1:0.1, 1:0.2, 1:0.3, and 1:0.4 were characterised by X-ray powder diffraction, Thermogravimetric analysis, fourier transform infrared spectrum and low temperature nitrogen adsorption measurements. According to the results, even a large quantity of silicon oxide component in the binary oxides system led to relatively small increase in defects in hexagonal lanthanum oxides obtained in a non-aqueous medium.
Graphical Abstract
La2O3–SiO2 binary oxide system was prepared by the sol–gel method without addition of strong polar solvents (e.g., water, alcohol). The present study is to propose a method for the synthesis of La2O3–SiO2 samples with the use of three components (tetraethoxysilane, acetic acid, lanthanum acetate). 
Similar content being viewed by others


1 Introduction
Lanthanum oxide has a lot of attractive properties that permitted its wide applications, e.g., as catalysts [1, 2], support for metal [3, 4], dielectric material [5, 6] and optical filters [7]. In our previous work [8, 9], we have studied the amorphous nature of SiO2–La2O3 system even for the largest quantity of lanthanum oxide (1:1 Si/La), which prefers crystalline forms. We have presented the influence of a specific type of anhydrous sol–gel synthesis on the products, besides we have determined such parameters such as thermal stability and textural analysis [10]. The study presented in this paper was undertaken for two reasons. The first aim was to study the structural changes in the crystalline lanthanum oxide with different amounts of the amorphous SiO2, and the second one was to determine the thermal stability and textural parameters of the samples studied. This study is a continuation of the investigation of La2O3-SiO2 binary oxides system applied as a support for metals catalysts with La/Si mole ratio between 1:0.0, and 1:0.4. The chemical stability of hexagonal La2O3 is insufficient, h–La2O3 usually reacts with H2O to form hydroxide lanthanum when it is left in natural air [11–13]. The presence of SiO2 component in the lanthanum oxide structure can modify the bulk and surface properties of this compound, which could be of key importance for consideration of binary oxide gel as a support for metallic catalysts with a crucial point of stabilisation of lanthanum oxide hydrolysis in the gel network [14]. Furthermore, the presence of amorphous SiO2 is expected to improve textural properties of the La2O3-SiO2 system. Traditional sol–gel methods for obtaining inorganic compounds are based on the hydrolysis-polycondensation reaction of mixed metal alkoxides, strong inorganic acids and water [13]. Particularly important problem is to control the reaction rate, which is very fast and leads to a reduction in microstructural monitoring of the final materials. An attractive solution is to use the sol–gel process without adding water to the initial system. The sol–gel technique in a water-free environment can lead to improved control of the hydrolysis reaction rate, essential for multicomponent systems in which the precursors applied differ much in the kinetics of hydrolysis [9]. By controlling the hydrolysis through a slow release of esterification water, we prevented formation of large aggregates of the half-products, and we could expect that the second component should be uniformly dispersed in binary oxide system as final product of synthesis. We proposed a new procedure for the synthesis of binary oxide gel systems using only three compounds: lanthanum acetate, tetraethylorthosilicate (TEOS), and AcOH in water free environment. This method permits a uniform distribution of the second reactant in the binary composite and makes it possible to design a new acid/base character of the surface of a catalyst or a support for metal catalysts.
2 Experimental
2.1 Synthesis of the sol–gel binary samples
A series of La2O3–SiO2 samples were synthesised by the sol–gel method, with La to Si molar ratio of: 1:0.0; 1:0.1; 1:0.2; 1:0.3 and 1:0.4. The samples were marked according to the increasing content of silicon as LaSi0.0, LaSi0.1, LaSi0.2, LaSi0.3 and LaSi0.4 respectively. The La2O3/SiO2 system was prepared on the basis of a modified procedure described in details in the previous paper [9]. The binary oxide gel system was obtained by mixing the appropriate amounts of TEOS and a solution of La(CH3COO)3 in anhydrous acetate acid. The homogenous solutions were closed in a reactor upon stirring and heated at 353 K for 48 h. The gels were dried at 393 K by slow evaporation for 72 h. Synthesised binary oxide gels were ignited on the evaporating dish, initiating a self-propagating combustion process, and dark grey loose product was obtained after the combustion was completed (Fig. S1 in Electronic Supporting Information). The LaSi0.0 sample was synthesised by decomposition of lanthanum acetate after evaporation of acetate acid under reduced pressure from the solution. All samples were placed in a muffle furnace, and preheated at 600 °C for 4 h. All products obtained were post-annealed in air atmosphere at 800 °C for 4 h.
2.2 X-ray powder diffraction (XRD) and rietveld refinement
XRD patterns were collected at room temperature by using a D8 Advance Diffractometer equipped with Johansson monochromator (λCu K α1 = 1,5406 Å) and silicon strip detector LynxEye. The intensities were measured in the 20–150o 2θ, with 0.02o step size and a measuring time of 2 s. per point. Crystalline phase identification was carried out using the diffractometer analytical software after revision of the PDF4+ (2013) 4.11.0.3 ICDD database crystalline compounds [15]. X-ray line profile analysis was used for microstructural characterisation of LaSi0.0 and LaSi0.4 samples. The standard line profiles for the instrumental line broadening evaluation were obtained from the diffraction pattern of the standard reference material LaB6 (SRM 660a) [16]. The Topas 4.2 programme was used for quantitative Rietveld phase analysis [17].
2.3 Thermogravimetric analysis (TGA)
TGA was carried out in SETSYS 12 (Setaram, France) thermoanalyzer. The sample (20 mg), in a alumina crucibles was heated in flowing air from room temperature to 1100 °C at the rate of 5 oC/min. For all experiments, we used the fraction of the grain sizes between 100 and 200 Mesh.
2.4 Fourier transform infrared spectrum (FTIR) spectroscopy
FTIR spectra were recorded with a BRUKER IFS88 spectrometer in the range of 4000–400 cm−1 with 128 scans and a resolution of 1 cm−1. The samples were prepared as pellets after dispersion in KBr matrix, and then the infrared transmission spectra were collected.
2.5 Porous structure
Textural parameters were obtained from the nitrogen adsorption–desorption isotherms at 77 K, and were recorded performed using Micromeritics ASAP V2.00D analyser. Prior to measurement, all samples were outgassed at 623 K at 0.4 Pa till constant weight. Both adsorption and desorption branches of the isotherm were taken in the p/p o range 0–1. The specific surface area was calculated using multipoint BET analysis and the pore sizes were measured by the Barrett–Joyner–Halenda (BJH) method of desorption.
3 Results and discussion
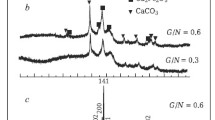
Figure 1 shows the XRD patterns of as-synthesised La/Si system by combustion method, and then calcined in a muffle furnace at 600 °C. All the diffraction peaks can be assigned to monoclinic phase with layered La2O2CO3-II compound [18]. G.A.M. Hussein has shown clearly that the thermal decomposition of lanthanum acetate hydrate near 500 °C leads to the formation of a more stable form of La2O2CO3 than La2(CO3)3 and La2O(CO3)2 [18]. The calculated lattice constants for the first sample LaSi0.0 are: a = 4.0807(7)Å, b = 13.506(3)Å, c = 7.0715(7) Å and β = 90.94(1)o, which are in good agreement with the reported values in PDF4+ ICDD database crystalline compounds (PDF 00-048-1113) [15]. The low intensity diffraction peaks indicate that the as-synthesised products have poorly crystallised, therefore a study of the effect of silicon incorporated in lanthanum oxycarbonate will be difficult. Additionally, the amorphous phase of SiO2 does not produce additional visible reflexes in the XRD diagrams. According to literature [19, 20] and the results of thermal analysis of the binary gel samples, the transformation of La2O2CO3 to La2O3 phase takes place. Figure 2 presents the results regarding the phase stabilities for LaSi0.0 and LaSi0.4. TG-TA-DTG curves for LaSi0.1, LaSi0.2, and LaSi0.3, respectively, are given in Fig. S2 in Electronic Supporting Information. The results obtained from the range of 300–1373 K indicate that all samples have only one endothermic distinct peak in the curves for the samples of each composition. Table 1 shows the decomposition temperatures, and compares the experimental and calculated weight losses in La2O2CO3 phases. The mass loss is attributed to the loss of carbon dioxide molecule and formation of lanthanum oxide: La2O2CO3 (s) ↔ La2O3 (s) + CO2 (g). The decomposition temperature slightly grows with increasing silicon content in La/Si system as a consequence of the slowdown of the decomposition process of La2O2CO3 surrounded by amorphous SiO2 in the bulk gel.

The XRPD patterns of La2O3/SiO2 samples calcined under air at 600 °C

TGA curves for LaSi0.0 and LaSi0.4 samples in the air atmosphere up to 1000 °C
The XRD patterns of La2O3-SiO2 samples calcined at 800 °C in air atmosphere are presented in Fig. 3. All the diffraction peaks can by assigned to hexagonal lanthanum oxide. Compared with La2O2CO3 phases (Fig. 1), the shape of the diffraction peaks of La2O3 phases (Fig. 3) is more clean-cut, and the diffraction intensity of La2O3 phases is stronger. Therefore, lanthanum oxide shows better crystalline quality than lanthanum oxycarbonate. The sample LaSi0.0 (La2O3) could be indexed on the basis of the hexagonal unit cell with the lattice parameters of a = 3.9375(2) Å, c = 6.1278(3) Å, and the space group of a P-3m1 [21, 22]. The unit-cell volume of all samples shows a linear increase with increasing SiO2 component in La2O3/SiO2 system (inset in Fig. 3). This expansion of unit cell volume (max 0.2%) can be explained by an increase in internal strains in a material, which can change the positions and shapes of x-ray diffraction peaks. With increasing silicon dioxide content in La/Si system, the diffraction peaks became broader, and their relative intensity decreases. It is well known that the defects could be caused by different sources: impurities, internal strains, planar faults, dislocations, or small precipitates. Crystals with defects are less precisely periodic in their atomic arrangements, but they still have distinct diffraction peaks. The peak broadening could be caused by both an increase in lattice strain and a reduction in crystalline size. Figure 4a shows the evolution of full width at half maximum (FWHM) peaks vs. 2θ for the first (LaSi0.0) and the last one (LaSi0.4) sample. Assuming that the strain is present, Williamson and Hall [23] have proposed a method for deconvoluting the size and strain broadening by treating the peak width as a function of 2θ. The strain is extracted from the slope, and the crystalline size is extracted from the y-intercept of the fit. Figure 4b shows the Williamson–Hall plots for LaSi0.0 and LaSi0.04 samples. The estimated values for the strain and crystalline size are given by 5.6755 × 10−4 (0.056%), 84 nm and 1.1033 × 10−3 (0.11%), 75 nm for LaSi0.0 and LaSi0.04, respectively. According to the results, the strain value increased and particle size decreased with increasing amorphous silicon dioxide content in the system.

The XRPD patterns of La2O3/SiO2 samples after sintering under air at 800 °C. Volume unit cell of La/Si system is given in the inset

The main aim of the high-temperature powder diffraction investigation was to determine the formation of types and proportional composition of lanthanum silicate oxides in the La/Si system. All samples were treated at 1200 °C (Fig. 5), and crystallised in three oxides: hexagonal lanthanum oxide [04-010-8687], monoclinic lanthanum silicon silicate oxide, La2SiO5, [04-010-8687] and hexagonal lanthanum silicate oxide La9.33(SiO4)6O2, [01-076-0340]. The quantitative Rietveld phase analysis for all samples is depicted in Fig. S3 in Electronic Supporting Information. The quantitative analysis results are summarised in Table 2. The results show clearly that the amount of hexagonal lanthanum oxide decreased with increasing silicon content in the oxides, in contrast to the behaviour for silicate oxides.

The Rietvled analysis results of X-ray diffraction phase identification of La2O3/SiO2 samples calcined under air at 1200 °C are given. The chemical compounds for phase identification are shown in the legend
FTIR of as-synthesised La/Si binary oxide gel system is shown in Fig. 6. The IR spectrum clearly confirmed the absence of chemical reaction between lanthanum oxide and silicon oxide, but on the other hand revealed strongly basic character of La2O3, it shows the band appearing at 3609 cm−1 that is attributed to the O–H stretching vibration groups from La(OH)3 [24]. The intensity of this peak decreases without shifting with increasing silicon in binary oxides gel. The peak at wavenumber 3452 cm−1 is assigned to the stretching vibrations of hydroxyl groups of molecular water on the oxide surface. In addition, the absorption bands at 2340, 1482, and 1388 cm−1 are attributed to the presence of carbonates [25]. Carbon dioxide and water react on the surface of lanthanum oxide under ambient conditions forming the chemisorbed surface carbonates and bicarbonates [26]. The characteristic peak seen at approximately 1090 cm−1 is assigned to the asymmetric stretching vibration of the three-dimensional Si–O–Si network [9]. As expected, the intensity of this band increases with increasing amounts the SiO2 component of La/Si gel. On the other hand, the FTIR spectra show a characteristic decrease in the intensities of the absorbance bands at about 643 and 865 cm−1 (La–O stretching vibration) with decreasing quantity of lanthanum oxide in the La/Si binary oxides gel system.

Position of the infrared bands as a function of concentration of silicon for gel samples in the La/Si system calcined at 800 °C
Low-temperature physical gas adsorption analysis is commonly used for determining the surface area and pore size distribution for the characterisation of a wide range of porous materials. Nitrogen physisorption measurements for LaSi0.0 and the LaSi0.4 are shown in Fig. 7. The pore size distributions and adsorption-desorption isotherm for all samples are presented in Fig. S4 in Electronic Supporting Information. All isotherms were type IV according to the IUPAC classification and the hysteresis loop type H3, typical of solids with slit-shaped pores. Type IV is an isotherm characteristic of a mesoporous solid and the hysteresis is due to capillary condensation into mesopores. Filling of mesopores took place at p/p 0 = 0.8–1.0 for all samples, except for the LiSi0.0 hysteresis, revealing tensile strength effect at p/p 0 = 0.4 leading to a forced closure of the hysteresis loop. The presence of narrowed mesopores causes delayed evaporation, particularly when the majority of those pores were narrowed to approximately 5 nm or lower [27, 28]. BJH pore size distribution with the corresponding mono-modal desorption pore diameters decrease (from 4.0 to 2.5 nm) with increasing SiO2 in the La/Si binary oxides system. On the other hand, the pore volume increases (from 0.0018 to 0.0036 cm3 g−1 nm−1) with increasing content of the SiO2 component. Results of the specific surface area, cumulative pore volume and the average pore diameters are presented in Table 3. The surface area and volume pore slightly increase with increasing concentration of the second oxide, in contrast to the average pore diameter, which is the effect of growth of defects on the surface of modified lanthanum oxide.

Comparison of pore size distributions for gels samples (LaSi0.00 and LaSi0.4) and associated adsorption-desorption isotherm with hysteresis loops
4 Conclusions
The textural and structural properties of crystalline La2O3/SiO2 oxides system prepared by non-aqueous sol–gel method were studied. The use of only three reactionary components permits obtaining simple sol–gel reaction system and allows the synthesis of organic-inorganic materials with new properties from ingredients insoluble in polar medium. The XRD patterns verified the imperfectness of hexagonal structure of lanthanum oxide during growth of crystals at amorphous silicon dioxide investigated in this work. An increase in the number of defects was noted with increasing silicon dioxide concentration in the studied system. The addition of amorphous SiO2 improves the textural properties of the gel surface. FTIR spectra data confirmed a decrease in the basic character of lanthanum oxide with increasing Si incorporation into the gel, which leads to a change in the hydrolytic properties of modified materials.
References
Rosynek MP, Magnuson DT (1977) Preparation and characterization of catalytic lanthanum oxide. J Catal 46:402–413
Ghiasi M, Malekzadeh A (2015) Synthesis, characterization and photocatalytic properties of lanthanum oxy-carbonate, lanthanum oxide and lanthanum hydroxide nanoparticles. Superlattices Microstruct 77:295–304
Hereijgers BP, Weckhuysen BM (2009) Selective oxidation of methanol to hydrogen over gold catalysts promoted by alkaline-earth-metal and lanthanum oxides. ChemSusChem 2(8):743–748
Nowicki W, Gąsowska A, Kirszensztejn P (2016) Investigation of interaction between the Pt(II) ions and aminosilane-modified silica surface in heterogeneous system. Appl Surf Sci 371:294–503
Wong H, Zhou J, Zhang J, Jin H, Kakushima K, Iwai H (2014) The interfaces of lanthanum oxide-based subnanometer EOT gate dielectrics. Nanoscale Res Lett 9(1):472–477
Zhao Y (2012) Design of higher-k and more stable rare earth oxides as gate dielectrics for advanced CMOS devices. Materials 5:1413–1438
Rieck JS, Bell AT (1986) Studies of the interactions of H2 and CO with PdSiO2 promoted with La2O3, CeO2, Pr6O11, Nd2O3, and Sm2O3. J Catal 99:278–292
Nowicki W, Rypka G, Kawałko A, Tolińska A, Kirszensztejn P (2014) Synthesis and characterization of SiO2-La2O3 gels obtained in a water-free enviroment. J Mater Sci 49:4416–4422
Kirszensztejn P, Kawałko A, Tolińska A, Przekop R (2011) Synthesis of SiO2–SnO2 gels in water free conditions. J Porous Mater 18:241–249
Bazzi R, Flores-Gonzales MA, Louis C, Lebbou K, Dujardin C, Brenier A, Zhang W, Tillement O, Bernstein E, Perriat P (2003) Synthesis and luminescent properties of sub-5-nm lanthanide oxides nanoparticles. J Lumin 102:445–450
Jothinathan E, Vanmeensel K, Vleugels J, Van der Biest O (2010) Synthesis of nano-crystalline apatite type electrolyte powders for solid oxide fuel cells. J Eur Ceram Soc 30:1699–1706
Rodrıguez-Reyna E, Fuentesa AF, Maczkab M, Hanuzab J, Boulahyad K, Amadore U (2006) Facile synthesis, characterization and electrical properties of apatite-type lanthanum germanates. Solid State Sci 8:168–177
Brinker CJ, Scherer GW (1990) Sol-gel science: the physics and chemistry sol–gel processing. Academic, New York, NY
Tanaka M, Sawaguchi T, Kuwahara M, Niwa O (2013) Surface modification of silicon oxide with trialkoxysilanes toward close-packed monolayer formation. Langmuir 29(21):6361–6368
JCPDS Powder Diffraction File International Centre for Diffraction Data. ICCD Powder diffraction database 2015-2016 PDF-4+. Newtown Square, PA, USA. 19073–3273
Cline JP, Deslattes RD, Staudenmann JL, Hudson LT, Henins A, Cheary RW (2000) NIST certificate, SRM 660a line position and line shape standard for powder diffraction. NIST, Gaithersburg, MD, pp 1–4
Bruker AXS (2008) TOPAS V4: general profile and structure analysis software for powder diffraction data. Bruker AXS, Karlsruhe
Olafsen A, Larsson AK, Fjellvåg H, Hauback BC (2001) On the crystal structure of Ln2O2CO3 II (Ln = La and Nd). J Solid State Chem 158:14–24
Hussein GAM (1994) Spectrothermal investigation of the decomposition course of lanthanum acetate hydrate. J Ther Anal 42:1091–1102
Bakiz B, Guinneton F, Arab M, Benlhachemi A, Gavarria J-R (2010) Elaboration, characterization of LaOHCO3, Ln2O2CO3, and La2O3 phases and their gas solid interactions with CH4 and CO gases. Moroc J Condens Matter 12(1):60–67
Hu C, Liu H, Dong W, Zhang Y, Bao G, Lao C, Wang ZL (2007) La(OH)3 and La2O3 nanobelts-synthesis and physical properties. Adv Mater 19:470–474
Salavati-Niasaria M, Hosseinzadeh G, Davar F (2011) Synthesis of lanthanum hydroxide and lanthanum oxide nanoparticles by sonochemical method. J Alloys Compd 509:4098–4103
Williamson GK, Hall WH (1953) X-ray line broadening from filed aluminium and wolfram. Acta Metall 1:22–31
Gangwar BP, Palakollu V, Singh A, Kanvah S, Sharma S (2014) Combustion synthesized La2O3 and La(OH)3: recyclable catalytic activity towards Knoevenagel and Hantzsch reactions. RSC Adv 4:55407–55416
Levan T, Che M, Tatibouet JM, Kermarec M (1993) Infrared study of the formation and stability of La2O2CO3 during the oxidative coupling of methane on La2O3. J Catal 142:18–26
Klingenberg B, Vannice MA (1996) Influence of pretreatment on lanthanum nitrate, carbonate, and oxide powders. Chem Mater 8:2755–2768
Voort P, Ravikovitch PI, Jong KP, Benjelloun M, Bavel E, Janssen AH, Neimark AV, Weckhuysen BM, Vansant EF (2002) A new templated ordered structure with combined micro- and mesopores and internal silica nanocapsules. J Phys Chem B 106:5873–5877
Groen JC, Peffer LAA, Pérez-Ramírez J (2003) Pore size determination in modified micro- and mesoporous materials. Pitfalls and limitations in gas adsorption data analysis. Microporous Mesoporous Mater 60:1–17
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Electronic supplementary material
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nowicki, W., Piskuła, Z.S., Kuźma, P. et al. Synthesis and characterization of a binary system La2O3–SiO2 prepared by combustion method. J Sol-Gel Sci Technol 82, 574–580 (2017). https://doi.org/10.1007/s10971-017-4317-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-017-4317-z