
Abstract
ZFAT is a transcriptional regulator, containing eighteen C2H2-type zinc-fingers and one AT-hook, involved in autoimmune thyroid disease, apoptosis, and immune-related cell survival. We determined the solution structures of the thirteen individual ZFAT zinc-fingers (ZF) and the tandemly arrayed zinc-fingers in the regions from ZF2 to ZF5, by NMR spectroscopy. ZFAT has eight uncommon bulged-out helix-containing zinc-fingers, and six of their structures (ZF4, ZF5, ZF6, ZF10, ZF11, and ZF13) were determined. The distribution patterns of the putative DNA-binding surface residues are different among the ZFAT zinc-fingers, suggesting the distinct DNA sequence preferences of the N-terminal and C-terminal zinc-fingers. Since ZFAT has three to five consecutive tandem zinc-fingers, which may cooperatively function as a unit, we also determined two tandemly arrayed zinc-finger structures, between ZF2 to ZF4 and ZF3 to ZF5. Our NMR spectroscopic analysis detected the interaction between ZF4 and ZF5, which are connected by an uncommon linker sequence, KKIK. The ZF4–ZF5 linker restrained the relative structural space between the two zinc-fingers in solution, unlike the other linker regions with determined structures, suggesting the involvement of the ZF4–ZF5 interfinger linker in the regulation of ZFAT function.
Similar content being viewed by others


Introduction
Autoimmune thyroid disease (AITD) is a general disease caused by the immune system responding to its own normal cells or organs, and to foreign antigens such as bacteria, viruses, and tumors [1–3]. The development of antibodies to antigenic thyroid components is a main feature of autoimmune diseases. ZFAT (Zinc finger gene in AITD susceptibility region; also known as ZNF406) was identified as a gene involved in the regulation of the autoimmune system [4]. The ZFAT protein is conserved from fish to human, and the human ZFAT protein is composed of eighteen C2H2-type zinc-fingers (ZFs) and one AT-hook motif between ZF1 and ZF2 [5] (Fig. 1a). ZFAT is expressed in peripheral B and T lymphocytes, and is also found in the human acute T lymphoblastic leukaemia cell line MOLT-4 and human umbilical vein endothelial cells [6, 7]. Notably, the ZFAT-knockdown in MOLT-4 induces apoptosis via the activation of caspases, suggesting that ZFAT is a transcriptional regulator involved in apoptosis and cell survival for immune-related cells. [6]. Furthermore, ZFAT is an essential transcriptional regulator for hematopoietic differentiation and indispensable for mouse embryonic development [8, 9], which indicates the critical role of ZFAT not only in AITD but also in a broad range of development and differentiation.

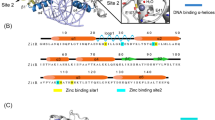
Primary structure of ZFAT. a Schematic representation of human ZFAT. The green and blue boxes indicate the C2H2 zinc-finger and the AT-hook motif, respectively. The positions of the zinc-fingers with solved structures are marked by asterisks (black human; violet mouse). b Sequence alignment of the ZFAT zinc-fingers. All of the human ZFAT zinc-fingers and mouse ZFAT zinc-fingers (mZF5 and mZF8 in violet) with solved structures are listed, with h and m indicating human and mouse, respectively. The zinc-coordinating Cys and His residues are colored cyan and magenta, respectively. The hash mark indicates the residues expected to be involved in DNA recognition. Secondary structures corresponding to the sequence are shown at the bottom
The transcriptional activity of ZFAT is considered to be mediated by its DNA-binding ZFs. The C2H2-type ZF, consisting of 20–30 residues, forms one N-terminal short antiparallel β-sheet and one helix [10]. The canonical C2H2 ZFs bind to specific DNA sequences, and the amino acids located at positions −1, +2, +3 and +6, from the N-terminal residue of the helix, directly contact a DNA base. The DNA recognition modes by these base-contacting residues were predicted from previous structural analyses, and the relationships between the base-contacting residues and the predicted DNA bases have been summarized as the recognition code [11]. In most cases, the C2H2-type ZF is repeated from two to more than thirty times in a protein [10]. Such tandem sets of ZFs are typically connected by a well-conserved TGEKP linker sequence [11–15]. These consecutive ZFs are known to bind to cognate DNA sequences as one functional unit [10, 16–19]. To understand the functional role of ZFAT in the regulation of the immune system, we determined the solution structures of single or consecutive tandem ZFs of ZFAT through an NMR method. We describe the structural features of the ZFAT ZFs, including the structural differences on the putative DNA recognition surfaces among the ZFAT ZFs, and the unique interaction mode within the tandem ZFs of ZF4 and ZF5, which are connected by an uncommon linker sequence.
Materials and methods
Protein expression and purification
The DNA sequences encoding the ZFs of the human and mouse ZFAT proteins (SwissProt accession numbers: Q9P243 and Q7TS63) were subcloned by PCR from the human and mouse cDNA clones by the two-step PCR method [20]. The individual domain regions used in this study are listed in Table 1. The cDNA fragments encoding these regions, along with those containing tandem ZF sequences, were cloned into the expression vector pCR2.1-TOPO (Invitrogen, Carlsbad, CA), as a fusion with an N-terminal poly-histidine affinity tag and a tobacco etch virus (TEV) protease cleavage site, and an artificial linker sequence (GSSGSSG) [20]. The actual sequences of the NMR samples contain these seven extra residues at their N-termini. The 13C/15N-labeled fusion proteins were synthesized by the cell-free protein expression system [21, 22], and were purified using a chelating column, as described previously [23, 24]. The purified proteins were concentrated to 0.1–1.2 mM in 20 mM Tris-d 11–HCl buffer (pH 7.0), containing 100 mM NaCl, 1 mM dithiothreitol-d 10, 50 μM ZnCl2, 1 mM iminodiacetic acid (IDA), 10 % D2O, and 0.02 % NaN3.
NMR spectroscopy and spectral assignments
All spectra were recorded on Bruker Avance 600, 700, 800, and 900 spectrometers at 296 or 298 K. Samples were first screened by 1H, 15N-HSQC spectroscopy [25]. The resonance assignments were accomplished using a conventional set of triple resonance spectra, as described previously [23, 24], and have been deposited in the Biological Magnetic Resonance data Bank (BMRB; Table 1). Inter-proton distance restraints were obtained from 15N and 13C edited NOESY spectra, both recorded with a mixing time of 80 ms. All spectra were processed using NMRPipe [26], and the programs Kujira [27] and NMRView [28] were employed for optimal visualization and spectral analyses.
Structure calculations
Automated NOE cross-peak assignments and structure calculations with torsion angle dynamics were performed using the software package CYANA [29, 30]. The backbone dihedral angle restraints from the TALOS program [31] were also included for the calculations, with allowed ranges of ±30°. The final structure calculations with CYANA were started from 100 conformers with random torsion angle values. The 20 conformers with the lowest final CYANA target function values were further refined with the AMBER12 program, using an Amber ff99SB force field and a generalized Born model, as described previously [32]. The tetrahedral zinc coordination was restrained by lower and upper distance limits, with force constants of 1000 kcal/mol/Å. All of the structures were validated using MolProbity [33, 34] and PROCHECK-NMR [35]. The structural statistics of the ZFAT ZFs are summarized in Supplemental Tables 1–3. Figures were generated with the MOLMOL [36] and PyMol (DeLano Scientific, San Carlos, CA) programs. All structures have been deposited in the Protein Data Bank. The PDB and BMRB accession codes of the structure-determined ZFAT ZF structures are provided in Table 1.
Results and discussion
Structural overview of the ZFAT zinc-fingers
The domain architecture of ZFAT is shown in Fig. 1a. The 1H, 15N and 13C assignments of each individual ZFAT ZF (Fig. 1b) expressed in the cell-free system were obtained by combining selected triple-resonance spectra. By screening the nature of the candidate protein samples, such as expression, solubility, and folding, we finally determined the following thirteen ZFAT ZF solution structures: human ZF2, ZF3, ZF4, ZF5, ZF6, ZF10, ZF11, ZF12, ZF13, ZF14 and ZF15; and mouse ZF5 and ZF8. All of the individual ZFs consisted of one N-terminal short antiparallel β-sheet and one helix (Figs. 1b, 2; Table 1), and their overall structures were similar to each other. On the other hand, the compositions of the putative DNA base-contacting surfaces differed among the solved zinc-finger structures (Fig. 3), suggesting functional divergence regarding their involvement and sequence specificity in DNA recognition.

Solution structures of isolated ZFAT zinc-fingers, depicted by ribbon diagrams. The zinc ion and the zinc-coordinating histidines and cysteines in each zinc-finger structure are colored yellow, magenta and cyan, respectively. The residues located at the positions that are potentially involved in DNA recognition are shown in green. The orientations of all of the structures are the same

Surface representation of isolated ZFAT zinc-fingers. Basic, acidic, and hydrophobic residues are colored blue, red, and yellow, respectively. The orientation of all of the structures is the same as in Fig. 2
Among the determined ZFAT ZF structures, six ZFs (i.e. hZF4, hZF5, mZF5, mZF8, hZF10, and hZF12) have a bulged-out helix structure, instead of a canonical helix structure (Fig. 4a). In a canonical helix, the zinc atom is held in a tetrahedral complex by the two Sγ of the C–X2–4–C sequence and the two Nε2 of the H–X3–H sequence, where C represents Cys, H is His, and X is any amino acid residue, and the subscript number represents the number of amino acid residues (Figs. 1b, 4a). On the other hand, the zinc atom in a bulged-out helix is held by the two Sγ of the C–X2–C sequence and the two Nε2 of the H–X4–H sequence (Figs. 1b, 4a). The bulged-out helix is amphipathic, with the side-chains of their hydrophobic faces packing the core of the domain and the exposed surface of the helix facing the hydrophilic residues involved in DNA recognition [37]. These structural features of the canonical or bulged-out helices are common among all of the ZFAT ZFs (Fig. 1b), and are also similar to those of other canonical C2H2 ZFs.

Structural features of ZFAT zinc-fingers. a Comparison of the bulged-out helix structure (hZF5, red) with that of the canonical helix (hZF2, blue). The zinc atom is depicted by a yellow ball. The zinc-coordinating Cys and His residues are depicted by cyan and magenta sticks, respectively. b Comparison of the loop structure of the C–X4–C type (hZF11, green) with that of the standard C–X2–C type zinc-finger (hZF12, blue). Other color codes are the same as in (a). The Cα positions of the protein–DNA complex structure of the DNA-binding zinc-finger of GLI (PDB ID: 2GLI) are used as the reference Cα positions. The position of the DNA (orange) is also from the GLI structure, for reference
C–X4–C type zinc-finger and bulged-out helix-containing zinc-fingers
There are two interesting structural features in the folds of the ZFAT ZFs. The first is that all of the ZFAT ZFs, except ZF11, have a short two-residue-spacer between the two zinc-coordinating cysteines, which is typically observed in Krüppel-type ZFs (i.e. C–X2–C). On the other hand, the ZF11 ZF has a long four-residue-spacer in the corresponding region (i.e. C–X4–C; Fig. 1b), yielding an extended β loop structure between the two antiparallel β strands (Fig. 4b). The N-terminal antiparallel β loop structure, which is formed by the interaction of zinc with the two zinc-coordinating cysteines, is essential for the stability of the overall ZF structure. When the zinc ion binds to an unfolded apo-form finger, it first interacts with the Cys residues and subsequently with the His residues [10]. The difference in the length between the two Cys residues is assumed to either modulate the stability or facilitate the interactions with other intramolecular ZFs [38]. The β loop structure may also function as a scaffold and affect the DNA-binding activity [39].
The second feature is that the ZFAT ZFs have an abundance of the abovementioned bulged-out helix structures (Figs. 1b, 4a). In the SMART database, (containing 274,117 C2H2 ZFs), approximately 80 % of the C2H2 ZFs (212,646) have the canonical H–X3–H motif, while only 15 % of the ZFs (40,767) have the H–X4–H motif. Notably, the ZFAT protein has eight bulged-out helix ZFs (44 %), including five with determined structures (i.e. hZF4, h/mZF5, mZF8, hZF10, and hZF12) and three putative bulged-out helix-containing ZFs (i.e. ZF1, ZF17, and ZF18), as judged from its amino acid sequence (Figs. 1b, 2). The percentage of bulged-out helix-containing ZFs of ZFAT, 44 %, is higher than that of frog TFIIIA (30 %), another known bulged-out helix ZF-containing protein [40]. The four-residue-spacing between the two histidines of the bulged-out helix, in which one amino acid is inserted into the canonical helix, is assumed to be critical for the structure and function of ZFAT. In order to maintain an ideal position for zinc coordination (see the two histidines in Fig. 4a), the additionally inserted residue causes the helix to bulge out slightly relative to those of the canonical helix ZFs. Consequently, this H–X4–H region forms a slightly larger and looser helical structure, as compared with the canonical H–X3–H helix, without distorting the overall ZF structure (Fig. 4a).
Although the backbone i–i-5 hydrogen bond, known as a π hydrogen bond, was formed between two His residues in each bulged-out helix, the backbone dihedral angles were quite different from those of the ideal π-helix (φ = −57.1; ψ = −69.7) [41, 42], as well as those of the ideal α helix (φ = −65.0; ψ = −40.0) [43, 44] (e.g., φ = −111.0 ± 16.1 for His349; and ψ = −36.6 ± 12.7 for Val348, respectively, in hZF4; see also Table 2). Therefore, as defined in the structural study of TFIIIA by Wuttke et al. [40], we used the term ‘bulged-out helix’ to describe an H–X4–H ZFAT ZF helix in this study, rather than the term ‘π-helix’. The unique bulged-out helix structure can allow distinct non-coordinating amino acids located in invariant positions to form hydrogen bonds with specific nucleotide bases in the major groove of DNA [10]. It also allows the canonical ZF helix, which is located adjacent to the bulged-out helix, to form extensive interactions with DNA [40].
Expected DNA recognition sequences of the ZFAT zinc-fingers
As for the molecular surfaces of the ZFAT ZFs, although all of the folds of the ZFAT ZFs are well conserved, the exposed surface of the helix for putative DNA-binding has a wide variety of physiochemical properties in the individual ZFAT ZFs (Fig. 3; see also Table 1). This suggested that some of the ZFAT ZFs contribute to the recognition of different DNA sequences or protein interactions [19]. In order to predict the DNA sequences recognized by the ZFAT ZFs, we applied the DNA-ZF recognition code [11], using our structural data. The possible DNA-contacting residues of the ZFAT ZFs at the key positions within the canonical or bulged-out helices (left panels), and the nucleotide preferred by each key residue of each ZF (right panels), are shown in Fig. 5. As for the bulged-out helix-containing ZFs, the residues involved in the extended interaction with DNA [40] are also shown in Fig. 5 (see the +10 residues in ZF4 and ZF17). From this prediction, it is plausible that the N-terminal half of the ZFAT ZFs may prefer DNA subsites containing AT-rich sequences (Fig. 5). This assumption is consistent with the fact that the AT-hook region prefers to bind an AT sequence existing between ZF1 and ZF2 (Fig. 1a). On the other hand, the C-terminal half of the ZFAT ZFs may prefer the DNA subsites containing GC-rich sequences (Fig. 5). Since consecutive ZFs bind to their corresponding DNA sequences in an anti-parallel fashion, where one ZF binds to one triplet DNA sequence and the adjacent C-terminal ZF binds to another triplet on the 5′-side [45, 46], the DNA sequence preferentially recognized by ZFAT may be a GC-rich sequence followed by an AT-rich sequence. Many C2H2 ZF proteins contain tandemly arrayed ZFs connected by specific linker sequences, while other members contain single or duplicated pairs of ZFs [10, 17]. Since the C2H2 ZFs are frequently involved in DNA-binding, variations in the numbers of ZFs and their spacing may affect DNA recognition [47]. Especially, multiple tandemly arrayed C2H2 ZFs can bind to the cognate DNA through two to three consecutive fingers [10, 16–19]. Based on the ZFAT domain architecture and the amino acid lengths of the linkers between the individual ZFAT ZFs, the following four ZFs are assumed to collaborate as DNA recognition units: [ZF2–ZF5] and [ZF6–ZF8] in the N-terminal half, and [ZF9–ZF12] and [ZF13–ZF17] in the C-terminal half (Fig. 1a). However, the precise target DNA sequence of ZFAT could not be identified, because of the lack of information about how these tandem ZF units cooperate with each other in recognizing a particular DNA sequence and how the bulged-out helix recognizes bases in a particular DNA sequence.

Expected DNA recognition sequences for each of the ZFAT zinc-fingers. The amino acid residues located at the key positions in the helix (−1, +2, +3, +6 and +10) of each zinc-finger are listed on the left, and the three to four base sequence (subsite) predicted to be preferred by each zinc-finger is shown on the right
Structural analysis of tandemly arrayed ZFAT zinc-fingers
In order to reveal the structural features of the tandemly arrayed ZFAT ZFs, we tried to determine the tertiary structures of tandem ZFAT ZF regions. We determined the solution structures of the tandem repeats ZF2–ZF3–ZF4 and ZF3–ZF4–ZF5 (Figs. 6a, 7; Table 1). The structures of the individual ZFs in the tandem ZF regions are quite similar to the corresponding isolated ZFs. Furthermore, the chemical shifts of almost all of the signals in both the tandem ZF regions and the isolated ZFs did not change, except for those detected in the terminal regions (data not shown). However, we found that the chemical shifts of the Ile352 (in the interfinger linker connecting ZF4 and ZF5) and Tyr330 (in ZF4) residues were quite different between the cases of the isolated ZF and the tandem ZF (Fig. 6b). Additionally, we observed several NOEs from Ile352 (in the interfinger linker connecting ZF4 and ZF5) to Tyr330 (in the β loop of ZF4), His349 (in the helix of ZF4), and Gln354 (in the β strand of ZF5). Although we could not determine the position of ZF5 relative to ZF4, because of the lack of clear interfinger NOEs between ZF4 and ZF5, these NOEs suggested that Ile352 may function as a clamp to limit the interdomain mobility between ZF4 and ZF5 (Figs. 6a, 7, 8a).

The uncommon interfinger linker reduces the flexibility. a The solution structures of the tandem ZF regions, ZF2–ZF3–ZF4 (top) and ZF3–ZF4–ZF5 (bottom). ZF2, ZF3, ZF4, and ZF5 are colored magenta, green, blue, and orange, respectively. In each structure, the central ZF is used for fitting. b Comparison of the 1H,13C HSQC spectra between ZF3–ZF4–ZF5 (top, black) and ZF5 (top, red), and ZF3–ZF4–ZF5 (bottom, black) and ZF4 (bottom, red). Signal assignments are labeled in the spectra

Comparison of interfinger flexibilities. The solution structures of the tandem ZF regions, ZF2–ZF3–ZF4 (top) and ZF3–ZF4–ZF5 (bottom), are shown. In each structure, the first, central, and last ZF-fitted superimposed structures are shown on the left, middle, and right, respectively. The color code is the same as in Fig. 6a

The role of the uncommon interfinger linker. a The solution structure of the tandem ZF3–ZF4–ZF5 region. ZF3, ZF4, and ZF5 are colored green, blue, and orange, respectively. The side chains for which inter-residue NOEs were observed are depicted by sticks. The observed NOEs are shown in yellow lines. b Comparison of the sequences of the interfinger linkers. The sequences of the interfinger linkers of ZFAT (ZF4–ZF5), Tramtrack, and a canonical C2H2 zinc-finger are listed
In contrast to the case with ZF4-ZF5, there were no such NOEs detected in ZF3–ZF4 and ZF2–ZF3. As a result, when the central ZF4 was fitted in the tandem ZF3–ZF4–ZF5, the RMSD and its standard deviation of ZF5 were both smaller than those of ZF3 (i.e. RMSD for ZF4-fitted ZF5 is 10.10 ± 4.82 Å, whereas that for ZF4-fitted ZF3 is 21.82 ± 9.62 Å; Figs. 6a, 7). Similarly, these values of the ZF4-fitted ZF5 in the tandem ZF3–ZF4–ZF5 were also smaller than those of both ZF3-fitted ZF2 and ZF3-fitted ZF4 in the tandem ZF2–ZF3–ZF4 (i.e. RMSDs for ZF3-fitted ZF2 = 15.25 ± 6.23 Å and ZF3-fitted ZF4 = 24.11 ± 9.01 Å; Figs. 6a, 7). Interestingly, previous linker mutation and NMR relaxation experiments revealed that the GAAP linker sequence of mouse testis zinc finger protein (TZFP) is more rigid than the canonical TGEKP linker sequence, lacking interfinger NOEs, in the absence of DNA [48]. Therefore, the linker sequence itself may affect the relative interfinger flexibility, without exhibiting interfinger NOEs.
The linker sequences between the canonical DNA-binding C2H2 ZFs are highly conserved, and are typically TGEKP. This sequence is necessary for DNA-binding and the interactions between two neighboring ZFs [11–15]. This canonical TGEKP linker is flexible in solution in the absence of DNA, whereas the linker in the DNA-bound complex forms a compact structure with a “snap-lock” helix-cap for stabilization of the DNA complex structure [13]. In addition, the TGEKP linker can be phosphorylated or acetylated, to regulate the DNA-binding activity of the tandemly arrayed C2H2 ZFs [49–51]. Intriguingly, this canonical linker sequence is not conserved in the several ZFAT interfinger regions. The linker sequence intervening between ZF4 and ZF5 is KKIK, which is completely different from the canonical linker sequence (Fig. 8b). In the case of the two tandem ZFs in Tramtrack, in which the linker sequence is KRNVKV (Fig. 8b), this linker is more flexible than the canonical TGEKP linker sequence, even upon DNA binding [52]. This flexibility reflected the absence of the helix-cap by the interfinger linker upon DNA binding and might contribute to the DNA binding mode where the DNA structure was distorted from the B form [13, 52].
In contrast, the structure of the KKIK linker between ZF4 and ZF5, which is another atypical linker sequence, was slightly restrained, even in the absence of DNA (Figs. 6a, 8). In the case of three tandemly repeats of TZFP which has the more rigid linker between ZF2 and ZF3, the mutation of the native GAAP linker to the canonical TGEKP linker obviously decreased the DNA binding of ZF3 [48]. The other interfinger linker sequences of ZFAT also differ from the highly conserved canonical TGEKP linker sequence, which may be related to the functions of the ZFAT ZFs in gene regulation. Further structural and biochemical analyses involving DNA-bound forms of ZFAT with tandem ZFAT ZFs, bulged-out helix-containing ZFs, and ZFAT interfinger linker sequences will be necessary to understand the molecular function of ZFAT.
Abbreviations
- AITD:
-
Autoimmune thyroid disease
- ZFAT:
-
Zinc-finger gene in AITD susceptibility region
- ZF:
-
Zinc-finger
- TEV:
-
Tobacco etch virus
- NMR:
-
Nuclear magnetic resonance
- HSQC:
-
Hetero-nuclear single quantum coherence
- NOE:
-
Nuclear Overhauser effect
- NOESY:
-
NOE spectroscopy
- PDB:
-
Protein Data Bank
- BMRB:
-
Biological Magnetic Resonance data Bank
References
Tomer Y, Davies TF (2003) Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev 24:694–717
Prummel MF, Strieder T, Wiersinga WM (2004) The environment and autoimmune thyroid diseases. Eur J Endocrinol 150:605–618
Jacobson EM, Tomer Y (2007) The CD40, CTLA-4, thyroglobulin, TSH receptor, and PTPN22 gene quintet and its contribution to thyroid autoimmunity: back to the future. J Autoimmun 28:85–98
Shirasawa S, Harada H, Furugaki K, Akamizu T, Ishikawa N, Ito K, Ito K, Tamai H, Kuma K, Kubota S, Hiratani H, Tsuchiya T, Baba I, Ishikawa M, Tanaka M, Sakai K, Aoki M, Yamamoto K, Sasazuki T (2004) SNPs in the promoter of a B cell-specific antisense transcript, SAS-ZFAT, determine susceptibility to autoimmune thyroid disease. Hum Mol Genet 13:2221–2231
Koyanagi M, Nakabayashi K, Fujimoto T, Gu N, Baba I, Takashima Y, Doi K, Harada H, Kato N, Sasazuki T, Shirasawa S (2008) ZFAT expression in B and T lymphocytes and identification of ZFAT-regulated genes. Genomics 91:451–457
Fujimoto T, Doi K, Koyanagi M, Tsunoda T, Takashima Y, Yoshida Y, Sasazuki T, Shirasawa S (2009) ZFAT is an antiapoptotic molecule and critical for cell survival in MOLT-4 cells. FEBS Lett 583:568–572
Yoshida Y, Tsunoda T, Takashima Y, Fujimoto T, Doi K, Sasazuki T, Kuroki M, Iwasaki A, Shirasawa S (2010) ZFAT is essential for endothelial cell assembly and the branch point formation of capillary-like structures in an angiogenesis model. Cell Mol Biol Lett 15:541–550
Tsunoda T, Takashima Y, Tanaka Y, Fujimoto T, Doi K, Hirose Y, Koyanagi M, Yoshida Y, Okamura T, Kuroki M, Sasazuki T, Shirasawa S (2010) Immune-related zinc finger gene ZFAT is an essential transcriptional regulator for hematopoietic differentiation in blood islands. Proc Natl Acad Sci USA 107:14199–14204
Doi K, Fujimoto T, Koyanagi M, Tsunoda T, Tanaka Y, Yoshida Y, Takashima Y, Kuroki M, Sasazuki T, Shirasawa S (2011) ZFAT is a critical molecule for cell survival in mouse embryonic fibroblasts. Cell Mol Biol Lett 16:89–100
Iuchi S, Kuldell N (2005) Zinc finger proteins: from atomic contact to cellular function. Kluwer Academic/Plenum Publishers, New York
Pabo CO, Peisach E, Grant RA (2001) Design and selection of novel Cys(2)His(2) zinc finger proteins. Annu Rev Biochem 70:313–340
Jacobs GH (1992) Determination of the base recognition positions of zinc fingers from sequence analysis. EMBO J 11:4507–4517
Laity JH, Dyson HJ, Wright PE (2000) DNA-induced alpha-helix capping in conserved linker sequences is a determinant of binding affinity in Cys(2)-His(2) zinc fingers. J Mol Biol 295:719–727
Wolfe SA, Nekludova L, Pabo CO (2000) DNA recognition by Cys(2)His(2) zinc finger proteins. Annu Rev Biophys Biomol Struct 29:183–212
Laity JH, Lee BM, Wright PE (2001) Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol 11:39–46
Nagaoka M, Kaji T, Imanishi M, Hori Y, Nomura W, Sugiura Y (2001) Multiconnection of identical zinc finger: implication for DNA binding affinity and unit modulation of the three zinc finger domain. Biochemistry 40:2932–2941
Iuchi S (2001) Three classes of C2H2 zinc finger proteins. Cell Mol Life Sci 58:625–635
Brown RS (2005) Zinc finger proteins: getting a grip on RNA. Curr Opin Struct Biol 15:94–98
Gamsjaeger R, Liew CK, Loughlin FE, Crossley M, Mackay JP (2007) Sticky fingers: zinc-fingers as protein-recognition motifs. Trends Biochem Sci 32:63–70
Yabuki T, Motoda Y, Hanada K, Nunokawa E, Saito M, Seki E, Inoue M, Kigawa T, Yokoyama S (2007) A robust two-step PCR method of template DNA production for high-throughput cell-free protein synthesis. J Struct Funct Genomics 4:173–191
Kigawa T, Yabuki T, Yoshida Y, Tsutsui M, Ito Y, Shibata T, Yokoyama S (1999) Cell-free production and stable-isotope labeling of milligram quantities of proteins. FEBS Lett 442:15–19
Kigawa T, Yabuki T, Matsuda N, Matsuda T, Nakajima R, Tanaka A, Yokoyama S (2004) Preparation of Escherichia coli cell extract for highly productive cell-free protein expression. J Struct Funct Genomics 5:63–68
Tochio N, Koshiba S, Kobayashi N, Inoue M, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Motoda Y, Kobayashi A, Tanaka A, Hayashizaki Y, Terada T, Shirouzu M, Kigawa T, Yokoyama S (2006) Solution structure of the kinase-associated domain 1 of mouse microtubule-associated protein/microtubule affinity-regulating kinase 3. Protein Sci 15:2534–2543
Tochio N, Umehara T, Koshiba S, Inoue M, Yabuki T, Aoki M, Seki E, Watanabe S, Tomo Y, Hanada M, Ikari M, Sato M, Terada T, Nagase T, Ohara O, Shirouzu M, Tanaka A, Kigawa T, Yokoyama S (2006) Solution structure of the SWIRM domain of human histone demethylase LSD1. Structure 14:457–468
Kigawa T, Inoue M, Aoki M, Matsuda T, Yabuki T, Seki E, Harada T, Watanabe S, Yokoyama S (2008) In: Spirin AS, Swartz JR (eds) Cell-free protein synthesis: methods and protocols. Wiley, New York
Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A (1995) NMRPIPE—a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR 6:277–293
Kobayashi N, Iwahara J, Koshiba S, Tomizawa T, Tochio N, Güntert P, Kigawa T, Yokoyama S (2007) KUJIRA, a package of integrated modules for systematic and interactive analysis of NMR data directed to high-throughput NMR structure studies. J Biomol NMR 39:31–52
Johnson BA, Blevins RA (1994) NMR view—a computer-program for the visualization and analysis of NMR data. J Biomol NMR 4:603–614
Güntert P (2004) Automated NMR structure calculation with CYANA. Methods Mol Biol 278:353–378
Jee J, Güntert P (2003) Influence of the completeness of chemical shift assignments on NMR structures obtained with automated NOE assignment. J Struct Funct Genomics 4:179–189
Cornilescu G, Delaglio F, Bax A (1999) Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR 13:289–302
Tsuda K, Someya T, Kuwasako K, Takahashi M, He F, Unzai S, Inoue M, Harada T, Watanabe S, Terada T, Kobayashi N, Shirouzu M, Kigawa T, Tanaka A, Sugano S, Güntert P, Yokoyama S, Muto Y (2011) Structural basis for the dual RNA-recognition modes of human Tra2-β RRM. Nucleic Acids Res 39:1538–1553
Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35:W375–W383
Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66:12–21
Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR 8:477–486
Koradi R, Billeter M, Wüthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14:29–32
Coleman JE (1992) Zinc proteins: enzymes, storage proteins, transcription factors, and replication proteins. Annu Rev Biochem 61:897–946
Berg JM, Shi YG (1996) The galvanization of biology: a growing appreciation for the roles of zinc. Science 271:1081–1085
Shiraishi Y, Imanishi M, Morisaki T, Sugiura Y (2005) Swapping of the beta-hairpin region between sp1 and GLI zinc fingers: significant role of the beta-hairpin region in DNA binding properties of C2H2-type zinc finger peptides. Biochemistry 44:2523–2528
Wuttke DS, Foster MP, Case DA, Gottesfeld JM, Wright PE (1997) Solution structure of the first three zinc fingers of TFIIIA bound to the cognate DNA sequence: determinants of affinity and sequence specificity. J Mol Biol 273:183–206
Low BW, Baybutt RB (1952) The pi-helix—a hydrogen bonded configuration of the polypeptide chain. J Am Chem Soc 74:5806–5807
Donohue J (1953) Hydrogen bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 39:470–478
Pauling L, Corey RB, Branson HR (1951) The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA 37:205–211
Pauling L, Corey RB (1951) Atomic coordinates and structure factors for two helical configurations of polypeptide chains. Proc Natl Acad Sci USA 37:235–240
Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO (1996) Zif268 protein-DNA complex refined at 1.6 Å: a model system for understanding zinc finger-DNA interactions. Structure 4:1171–1180
Elrod-Erickson M, Benson TE, Pabo CO (1998) High-resolution structures of variant Zif268-DNA complexes: implications for understanding zinc finger-DNA recognition. Structure 6:451–464
Nagaoka M, Nomura W, Shiraishi Y, Sugiura Y (2001) Significant effect of linker sequence on DNA recognition by multi-zinc finger protein. Biochem Biophys Res Commun 282:1001–1007
Chou CC, Lou YC, Tang TK, Chen C (2010) Structure and DNA binding characteristics of the three-Cys(2)His(2) domain of mouse testis zinc finger protein. Proteins 78:2202–2212
Dovat S, Ronni T, Russell D, Ferrini R, Cobb BS, Smale ST (2002) A common mechanism for mitotic inactivation of C2H2 zinc finger DNA-binding domains. Genes Dev 16:2985–2990
Song CZ, Keller K, Chen YC, Stamatoyannopoulos G (2003) Functional interplay between CBP and PCAF in acetylation and regulation of transcription factor KLF13 activity. J Mol Biol 329:207–215
Jantz D, Berg JM (2004) Reduction in DNA-binding affinity of CYS(2)His(2) zinc finger proteins by linker phosphorylation. Proc Natl Acad Sci USA 101:7589–7593
Fairall L, Schwabe JW, Chapman L, Finch JT, Rhodes D (1993) The crystal structure of a two zinc-finger peptide reveals an extension to the rules for zinc-finger/DNA recognition. Nature 366:483–487
Acknowledgments
We thank Ms. Tomoko Nakayama, Ms. Takako Imada, and Mr. Kojiro Ake for clerical assistance. This work was supported in part by the RIKEN Structural Genomics/Proteomics Initiative (RSGI) in the National Project on Protein Structural and Functional Analyses, from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, and the Platform for Dynamic Approaches to Living System of MEXT.
Author information
Authors and Affiliations
Corresponding author
Additional information
Naoya Tochio and Takashi Umehara have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Tochio, N., Umehara, T., Nakabayashi, K. et al. Solution structures of the DNA-binding domains of immune-related zinc-finger protein ZFAT. J Struct Funct Genomics 16, 55–65 (2015). https://doi.org/10.1007/s10969-015-9196-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10969-015-9196-3