
Abstract
Ruthenium is a fission product that can be released from the fuel in case of a severe nuclear accident. In this work the impact of the atmosphere composition, including air radiolysis products, on the transport of ruthenium through a primary circuit was examined. Experiments were performed at temperatures 1300, 1500 and 1700 K in a slightly humid air. In the experiments significant effect of nitrogen oxides (N2O, NO2) and nitric acid on the ruthenium chemistry in the model primary circuit was observed. The obtained results indicate a strong effect of air radiolysis products on the quantity partitioning of transported ruthenium to gaseous and aerosol compounds.
Similar content being viewed by others
Introduction
In a nuclear accident the main concern is that elements prone to form volatile compounds will be released from the fuel. Due to the ability of ruthenium to form volatile oxides and radiological risk via isotopes 103Ru and 106Ru, ruthenium is concerned as one of the critical elements in the case of a nuclear accident.
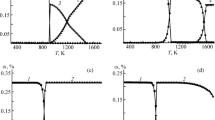
Proper quantification of the release and transport rates of radionuclides is necessary to evaluate the possible source term as accurately as possible. Consequent interactions of these nuclides with surface materials within the containment and other fission products can be then evaluated. The release of fission products from the irradiated nuclear fuel samples under different experimental conditions was investigated during the PHÉBUS FP and VERCORS research programs [1–3]. Under these integral experiments releases of ruthenium were significant when up to 17% of ruthenium content was released from the fuel [1]. A strong dependence of the ruthenium release rates on the oxygen content and temperature was observed [2, 3]. The thermodynamic equilibrium composition of ruthenium gaseous species calculated by the Factsage thermochemical software predicts the main volatile oxides to be RuO2, RuO3 and RuO4 depending on temperature, as shown in Fig. 1 [4].

Ruthenium gaseous species at thermodynamic equilibrium in air atmosphere at 1 bar pressure [4]
Both gaseous RuO2 and RuO3 are unstable at lower temperatures, which makes their partial pressures in the gaseous atmosphere very low at temperatures below 1000 K [5]. RuO2 condenses and RuO3 decomposes into the form of solid RuO2 according to Eqs. (1) and (2), where K is the equilibrium constant [5]. Although RuO4 is also not stable at the low temperatures, its decomposition kinetics are much slower when compared to RuO3(g) and RuO2(g) [6]. Thus RuO4 is the most relevant form in the conditions where temperatures lower than 1000 K are expected.
Very few studies deal with the transport of ruthenium through the primary circuit of a nuclear power plant [7–9]. The humidity, temperature and the flow rate of air-flow have been shown to be the main factors affecting the ruthenium transport in the primary circuit conditions. The transported ruthenium was observed to be in the form of aerosols consisting of RuO2 and in the form of gaseous RuO4. The quantity and the chemical form of transported ruthenium can also be affected by interactions with other elements released from the fuel [10]. Thus using the pristine air atmosphere in the experiments seems to be oversimplified in the case of severe accident studies due to the occurrence of aerosols (e.g., fission products, control rod materials) as well as various gaseous compounds, such as produced by the radiolysis of air [1, 11]. Experimental data is lacking, however, for atmospheric compositions other than dry or humid air. Only a few studies have addressed the effect of gaseous and particulate additives on the ruthenium chemistry in air–steam atmospheres [10, 12–15].
During a nuclear accident the main air radiolysis products expected in the containment atmosphere are ozone and nitrogen oxides such as NO2 and N2O. When humid atmosphere is taken into account the reaction of NO2 with water leads to the production of HNO3. All these compounds show oxidizing properties [16] and thus can oxidize lower ruthenium oxides into the form of RuO4 in the primary circuit of a nuclear power plant. The proposed reactions of RuO2 and RuO3 oxidation to RuO4 are presented in Eqs. (3)–(8) together with the corresponding equilibrium constants as calculated by HSC 5.11 chemistry software [17].
To provide a more precise and realistic modelling of the ruthenium chemistry in the primary circuit conditions the interaction of aerosols and air radiolysis products with Ru oxides in the gas phase needs to be evaluated. Therefore, to have a better insight into the chemistry of ruthenium during its transport through the RCS, the effects of nitrogen compounds (NO2, N2O, HNO3) on the transport and speciation of ruthenium were examined in this work.
Experimental
Experimental facility and procedure
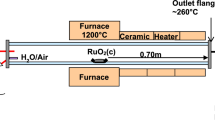
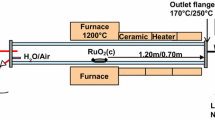
The configuration of the “VTT’s Ru transport facility” for the experiments is presented in Fig. 2. A detailed description of the facility is provided in previous work [7, 13, 15]. The main component of the facility was the horizontal, tubular flow furnace (Entech, ETF20/18-II-L). This was used to heat the anhydrous RuO2 powder (99.95%, Alfa Aesar). The furnace was 110 cm long and had two heating sections, each 40 cm long. These zones were separated by a 38 mm layer of insulation. At both ends of the furnace there was 131 mm of thermal insulation. The furnace tube was made of high purity alumina (Al2O3, 99.7%) and its inner diameter was 22 mm. The alumina crucible (length 20 cm) with the RuO2 powder (mass 1 or 2 g depending on the temperature used in the experiment) was placed at the beginning of the second heated zone of the furnace. As a new feature in these experiments a second alumina tube (Al2O3, 99.7%, outer diameter 6 mm with a wall thickness of 1 mm) was inserted inside the furnace tube, with the outlet located directly after the crucible. The RuO2 powder was heated to 1300, 1500 or 1700 K in an oxidizing flow in order to produce gaseous ruthenium oxides.

Schematics of the experimental facility for ruthenium transport studies
The total flow rate through the facility was 5.0 ± 0.1 l/min (NTP; conditions 0 °C, 101,325 Pa, measured using a thermal mass flowmeter TSI 3063, TSI Incorp.). Half of the total flow was directed through the inner furnace tube and the rest of the flow passed through the furnace tube. The pressure inside the facility ranged from 102 to 104 kPa. The air flow (2.5 ± 0.1 l/min, NTP) directed to the furnace tube was fed through an atomizer (TSI 3076). The air flow transported the water droplets (Milli-Q, ultrapure water, resistivity of 18.2 MΩ cm at 25 °C) produced by atomizer via the heated line (120 °C) into the inlet of the furnace. Water evaporated when the droplets were heated and therefore led to an increase in the steam concentration within the furnace. A flow of N2O, NO2 or HNO3 gases (2.5 ± 0.1 l/min, NTP) was fed through the inner furnace tube. NO2 was diluted with N2 to obtain a similar concentration of precursor as in the case of N2O. As HNO3 was fed using an additional atomizer located before the inlet of inner furnace tube (not shown in Fig. 2), a carrier gas of nitrogen was used to transport HNO3 droplets (solution of HNO3 and Milli-Q water) via the heated line (120 °C) into the inlet of the inner furnace tube. The experimental matrix is presented in Table 1. The duration of experiments was 60 min for the experiments conducted in the humid air atmosphere (experiments 1–3) and 20 min for the experiments with additive precursors fed into the humid air atmosphere (experiments 4–12).
After the vaporization of Ru and the following reactions within the gaseous atmosphere, the gaseous and particulate reaction products were trapped in a NaOH solution and collected on planer filters, respectively. Further details of this are provided in a previous work [13]. Particles were also analyzed online (see details below).
Analysis methods
Ruthenium release
The release rate of ruthenium in the experiments was obtained by weighing the mass of the crucible containing RuO2 before and after the experiments. The mass of released RuO2 was then converted to the corresponding mass of elemental ruthenium. Based on the previous study performed with the same facility using 103Ru radiotracer [7], the release of ruthenium from the crucible was assumed to be linear during the experiments.
Online analysis of ruthenium transport
The number size distribution of particles was measured online with a combination of a differential mobility analyser (DMA, TSI 3080/3081) and a condensation particle counter (CPC, TSI 3775) with a time resolution of 3 min. The flow rate through these devices was 0.30 ± 0.01 l/min (NTP). The particles were size classified according to their electrical mobility by the DMA and the number of particles in each size class was counted by the CPC (with a counting efficiency higher than 96%). The measurement range was from 15 to 670 nm. However, a pre-impactor removed particles larger than 615 nm at the inlet of the DMA. The measurement system was controlled with the Aerosol Instrument Manager software version 9.0 (TSI). This measurement system is known as a scanning mobility particle sizer (SMPS).
All the online measurement data presented was corrected by considering the loading of the analysis filter by particles and the following decrease in the flow rate through the filter, and therefore took into consideration the decreased flow rate into the aerosol sampling line from the main line. Correction was based on the calibration of flow rate through the critical orifice (CO) at various temperatures and pressures, simulating the loading of the filter. The calibration data was then used to estimate the flow rate of CO in the experiments, with the help of temperature and pressure measurement data. The flow rate from the main line to the aerosol line was also always measured with a thermal mass flowmeter at the beginning of every experiment. As a result, the changes in dilution ratio could be taken into consideration. The highest uncertainty in the dilution ratio originated from the inaccuracy of the mass flow controller feeding air through the porous tube dilutor and the thermal mass flowmeter. Given that the uncertainty of both devices can be ±2% of the reading, the uncertainty in the dilution ratio was ca. ±4%. Otherwise the contribution of uncertainties in temperature and pressure measurements to the dilution ratio was low, since the flow rate through the CO did not vary significantly due to these uncertainties. The online data presented was also dependent on the flow rate through the main line. The flow rate was always measured at the beginning of the experiments, and an additional uncertainty of ±2% resulted from the flowmeter. Therefore, the combined conservative uncertainty estimate for the online data presented was ca. ±6%. The particle number concentration values measured with CPC could also be too low by up to 4% due to the deficiency in counting efficiency.
Instrumental neutron activation analysis (INAA)
The quantification of ruthenium aerosols collected on filters and gaseous ruthenium trapped in the sodium hydroxide liquid traps was carried out by INAA. Ruthenium in the liquid traps was precipitated with addition of EtOH (96% Sigma-Aldrich), centrifuged and then filtered from the solution. Aerosols collected on the PTFE filters were used as they were after the experiment. Samples were then irradiated in the research reactor at VTT (Triga mark II reactor in Otaniemi, Espoo). Irradiations were performed with a thermal neutron flux of 8.7 × 1012 n/cm2/s and epithermal flux of 4.6 × 1012 n/cm2/s. Samples were irradiated for periods of from 10 min up to 4 h, depending on the ruthenium content in the sample. After 1 week of cooling time, the samples were measured by means of gamma spectrometry.
For these measurements a high purity germanium detector (Ortec model GEM-15180-S) was used with a relative efficiency of 17.7% and resolutions of 1.7 keV at 1332 keV. The evaluation of data was carried out using GammaVision software version 7.01.03. (Ortec). The detector was empirically calibrated for both energy and efficiency with QCYA18189 (Eckert and Ziegler) standard radionuclide source solution with the same geometry as irradiated samples.
The activity of 103Ru was determined from counts at the 497 keV peak, where absolute efficiency at a given geometry was determined to be 1.7%. The detection limit for ruthenium was determined to be 1.0E−2 µg based on the times of irradiations and measurements. Cross sections used for the data evaluation were taken from a previous publication [18]. Uncertainty of the measurements was calculated to be 5% according to the guide to the expression of uncertainties in measurements [19].
Chemical characterization
X-ray photoelectron spectroscopy
The collected solid samples were analyzed using X-ray photoelectron spectroscopy (XPS) to obtain chemical characterization of the aerosols. For the XPS measurements a Perkin Elmer Phi 5500 multi technique system was used. The detailed setup of the machine during measurements was described in a previous work [20]. Commonly, the C 1s peak originating from the unavoidable atmospheric contamination is used as an internal standard for the binding energies (BEs) during XPS measurements. In the case of ruthenium the Ru 3d5/2 peak and the C 1s peak are overlapping, making this reference unreliable. To avoid this problem the gold foil conductively connected to the measured samples was used as an internal standard during the measurements. The experimental uncertainty of BE of the Ru 3d5/2 peak was determined to be ±0.1 eV. The curve fitting of the obtained spectra was made using PHI Multipak software (Ulvac-Phi, Inc.), assuming Shirley background. The asymmetrical shape of peaks was used due to the conductive nature of anhydrous RuO2 [21]. XPS analysis was performed using at least two different spots on the samples.
X-ray diffraction analysis (XRD)
Crystallographic structures of the collected aerosols were examined by XRD analysis. The combination of XPS and XRD analysis allowed the characterization of both crystalline and potentially amorphous compounds in the collected aerosols. XRD measurements were made using a Bruker D2 Phaser diffractometer with Cu Kα characteristic radiation, equipped with a scintillation detector. Rotation speed of the sample holder was 360°/min and the measurement angle interval was 20°–80° 2θ. The comparison of the obtained data with standards in the Joint Committee of Powder Diffraction Standards database [22] led to the crystal structure identification of the collected compounds.
Results
Release and transport results
Release of ruthenium
The amount of the released ruthenium from the crucible was obtained as the mass difference of the crucible with RuO2 precursor before and after the experiment. The obtained release rates are presented in Table 2.
As can be seen from Table 2 the release rates of ruthenium increased with increased temperature. As the location of the injection of additional precursors into the airflow was just after the crucible, the precursors did not affect the vaporization of ruthenium and the observed ruthenium release results were as expected. When compared with the previous experiments the decrease in airflow over the crucible from 5.0 to 2.5 l/min resulted in an approx. 50% decrease of the ruthenium release rate from the crucible [7, 13]. This effect can be attributed to the lower absolute amount of oxygen available for the oxidation of ruthenium from the crucible.
Ruthenium transport
The quantities of transported ruthenium both in the form of aerosols and gaseous ruthenium trapped in sodium hydroxide traps were obtained by means of neutron activation and consequent gamma spectroscopy measurements. The obtained quantities, presented as % of released ruthenium, are shown in Table 3.
After each experiment a significant amount of ruthenium was visually observed to be deposited at the outlet of the furnace, where the temperature gradient was the steepest. Similar behavior was observed in the previous work using the same facility [23]. The separate effects of different precursors are discussed in subsequent sections.
Air atmosphere
The masses of ruthenium transported in gaseous and aerosol forms in the humid air atmosphere were determined and are presented in Table 4. From Table 4 it can be seen that the aerosol form predominated over RuO4 over the entire temperature interval of the experiments (1300–1700 K). The increase of the temperature in the experiments decreased the transported fraction of gaseous ruthenium through the facility. This observation is in agreement with the trend in thermodynamic equilibrium calculations performed with the HSC 5.11 software [17] as well as with the trend presented in Fig. 1. The increased temperature in the experiments led to increased overall transport of ruthenium through the facility. When data from this work was compared with the previous experiments [13] the gaseous fraction of ruthenium transported through the facility was lower. This may indicate the effect of the flow rate on the transport of RuO4 through the RCS. A similar effect was observed in the study of Vér et al. [9] where very low flow rates were used.
Atmosphere with 50 ppmV NO2
The results of ruthenium transport experiments under a humid air atmosphere with 50 ppmV of NO2 additive are shown in Table 5. The introduction of NO2 into the airflow had a significant effect on the composition of transported ruthenium. At temperatures of 1300 and 1500 K the fraction of the gaseous ruthenium transported through the facility was strongly increased when compared to the experiments in the humid air atmosphere at the same temperature.
Over the entire experimental temperature range a strong increase of the transported gaseous ruthenium fraction and corresponding decrease of aerosol was observed when compared with the humid air atmosphere experiments. This behaviour can be explained by the oxidation of RuO3(g) in the hot zone of the furnace according to reaction (9). The equilibrium constants for reaction (9) were calculated using the HSC 5.11 software [17] and these values are presented in Table 6. The ratios between the transported aerosol and gaseous fractions of ruthenium as presented in Table 5 lower than thermodynamic equilibrium calculations predict (Table 6) in experiments 5 and 6 and higher in experiment 3.
Additionally, as can be seen from Table 5, the fraction of gaseous ruthenium transported through the facility decreased with increasing temperature. This effect was attributed to two different phenomena; firstly the thermal decomposition of NO2 at high temperatures [24] according to reaction (10) [25]
and secondly the decreasing ability of NO2 to oxidize RuO3(g) to RuO4(g) as presented in reaction (9), with a temperature increase according to the equilibrium constants presented in Table 6.
As can be seen from the data in Tables 4 and 5, the total amount of transported ruthenium increased over the entire temperature range when compared to the humid air atmosphere.
Atmosphere with 50 ppmV N2O
The obtained amounts of ruthenium transported through the facility with injection of N2O gas are presented in Table 7. The injection of N2O increased the transported aerosol fraction of ruthenium when compared to the humid air experiments. This behavior was partly attributed to reactions (11) and (12) and the subsequent decomposition of RuO3 into solid RuO2 at the outlet of the hot zone from the furnace, where a temperature decrease below 1000 K was observed.
The amount of total ruthenium transported showed strong temperature dependence behavior. At 1300 K there was a decrease in the total amount of transported ruthenium in comparison to the humid air atmosphere. At 1500 K the total amount of transported ruthenium was almost double that observed in humid air. At 1700 K the observed increase of ruthenium transport due to NO2 injection was statistically insignificant when compared to the humid air experiments.
Atmosphere with 5 ppmV HNO3
The quantities of ruthenium transported in an atmosphere with 5 ppmV HNO3 are presented in Table 8. The introduction of HNO3 into the airflow resulted in a higher gaseous fraction of ruthenium being transported through the facility when compared to the humid air atmosphere. This effect was observed over the entire temperature range used in the experiments.
As can be seen from Tables 3 and 8 the effect of nitric acid was not as prominent as predicted by the thermodynamic calculations. These, calculated using HSC 5.11 software, indicated that K values for reaction (13) would be 1.65E11, 4.57E10 and 1.66E10 for temperatures 1300, 1500 and 1700 K, respectively [17]. This observation can again be explained by the thermal decomposition of HNO3 to the lower nitrogen oxides [26, 27] at elevated temperatures, thus lowering the amount of precursor in the gas phase.
When the values in Tables 4 and 8 were compared, the total amount of transported ruthenium was fairly similar when compared to the humid air atmosphere over the entire temperature range used in the experiments.
Online monitoring of aerosol transport
In order to understand the transient behavior of ruthenium, the transport of aerosol particles through the facility was followed online. The number concentration, diameter and number size distribution of particles were measured with SMPS at the outlet of the facility. The range of measurement uncertainty (±10% in the experiments) is not displayed in Fig. 3 or 4. The data for experiment 10 is not presented as there was a fault in the online measurement.

The particle number concentration (#/cm3, above) and count median diameter (nm, below) at the outlet of the facility during the experiments (measured with SMPS). The duration of experiments 1–3 was 60 min, whereas the other experiments lasted for 20 min

The particle number size distribution at the time point of 750 s since the beginning of each experiment (measured with SMPS)
In Fig. 3, the development of particle number concentration and the count median diameter (CMD) of particles over the course of the experiments are presented. On the basis of the measurement results, the number concentration of particles remained at a rather similar level in the experiments when only the effect of RuO2 vaporization temperature was examined. However, the effect of vaporization temperature on the diameter of particles was significant. The temperature increase from 1300 to 1700 K caused an increase in particle diameter in every experiment, resulting in up to 3.5 times larger particles in the case of NO2 feed. This phenomenon is directly connected to a higher release of ruthenium from the crucible and to the subsequent formation of particles. High release of ruthenium also favors the agglomeration of particles when the concentration of particles exceeds ca. 106 particles/cm3 [28].
The transport of particles was affected by the feed of nitrogen compounds (NO2, N2O, HNO3) into the flow of Ru oxides when compared with reference experiments 1–3. In general, the number concentration of particles decreased, but at the same time the diameter of particles seemed to increase. Depending on the experiment, the particle CMD ranged from ca. 20 to 210 nm. In the case of NO2 feed, the measured particle concentration was at the lowest level, ranging mainly from ca. 103 to 106 particles/cm3 in experiments 4 and 5. The concentration increased in experiment 6 and was observed to be between ca. 106 and 107 particles/cm3. Furthermore, the particle diameter was also greatest in experiment 6 and seemed to even increase strongly over the course of the experiment. This indicates, in addition to the agglomeration of particles, that part of the formed gaseous Ru compounds were probably condensing on the surface of the existing particles, thereby increasing the particle diameter. This conclusion is also supported by the low number concentration of particles measured and the previous observation of high formation of gaseous Ru due to NO2, see [13]. The effects of N2O and HNO3 feeds on the particle properties were not as strong as the NO2 feed. Therefore, the observed effects on the particle number concentration and particle diameter were in between the range limited by the reference experiments and NO2 experiments (see above).
The particle number size distribution for a particle diameter range from 15 to 500 nm in the experiments is presented in Fig. 4. The data is presented at the time point of 750 s since the beginning of each experiment. In addition to the above observations on particle behavior, it was noticed that the transported particles were lognormally distributed and that most of the particles were smaller than 500 nm in diameter. The feed of nitrogen compounds N2O and HNO3 under the studied conditions did not vary the shape of the particle number size distribution greatly. The broad particle distribution and the predominance of large particles (100–500 nm) in the distribution were evident when NO2 was present in the atmosphere, see for example the case of 1700 K.
Chemical characterization
XPS analysis
The aerosol particles transported through the facility were collected on PTFE filters and then examined with XPS. With the XPS technique the BEs of electrons in the elements of interest could be determined. The identification of chemical composition was obtained by comparing determined BE values with the reference values from the literature. In the cases of anhydrous and hydrated RuO2 references commercial powders (purity 99.5%, Alfa Aesar) were analyzed in-house and the obtained reference spectra were then compared with the spectra of the collected aerosols.
A selection of the reference BE values used during the evaluation are presented in Table 9. From the values in Table 9 it is clear that the BE is not only dependent on the oxidation state of ruthenium but also on the chemical environment, e.g., the hydration of RuO2. Similar observations were also made in a previous study [29].
The BEs of the Ru 3d5/2 peak in all samples were determined to be within the interval of 280.4–280.5 eV, as presented in Fig. 5. This value provides a very good fit with the Ru 3d5/2 BE in the anhydrous form of RuO2, thus indicating that the form of ruthenium in the transported aerosol was anhydrous RuO2 under all experimental conditions. The overall characteristics of the spectra are very similar to each other, therefore strengthening the assumption that all obtained spectra originate from the same compound.

The XPS spectra obtained from measurements of aerosols collected on PTFE filters. Spectra were scaled to fit the figure
Nitrogen was not detected in the collected aerosol samples. The possible formation of ruthenium nitrosyl compounds [30, 31] could therefore be ruled out during the data evaluation process.
XRD
The spectra obtained from the qualitative crystallographic XRD analysis of the collected aerosol samples are presented in Fig. 6. The XRD spectra recorded from experiments 1 to 12 showed the same diffraction pattern, which corresponds to the rutile structure of RuO2. This is in good agreement with the XPS analysis, leading to the conclusion that aerosols collected from the gas flow were in the form of anhydrous ruthenium dioxide.

The XRD spectra obtained from the samples in experiments 1–12. The height of the peaks was scaled in order to fit in the figure
Conclusions
The main objective of this study was to evaluate the effects of different nitrogen compounds on the transport and chemical composition of ruthenium in a model primary circuit of NPP. Nitrogen oxides and HNO3 represented the air radiolysis products unavoidably formed during a nuclear accident connected with air ingress sequence. The experiments were performed at temperatures of 1300, 1500 and 1700 K, with air atmosphere to simulate an air ingress type of accident. The examination of the quantities and chemical composition of transported ruthenium both in aerosol and gaseous form through the primary circuit simulating facility was within the scope of the study.
The effects of humid air, NO2, N2O and HNO3 on the transport and partitioning of ruthenium were investigated in this work.
It was shown that the release rate of ruthenium (given as for elemental Ru) from the ruthenium dioxide powder was strongly dependent on the temperature of the experiment. The determined release rate values were 0.34 ± 0.07 mg/min at 1300 K, 3.22 ± 0.16 mg/min at 1500 K and 20.27 ± 1.04 mg/min at 1700 K in an air atmosphere with a low steam content (≈2.1E4 ppmV).
The quantification and partitioning of ruthenium was obtained by collection of aerosols on PTFE filters and trapping of the gaseous fraction in 1 M NaOH solution. It was visually observed that the major part of the released ruthenium was deposited within the area of the furnace outlet where the temperature gradient was the steepest. It was also shown that the temperature increase during the experiments increased not only the release but also the transport of ruthenium through the facility. The quantification of ruthenium transport demonstrated a significant impact of the gaseous additives on both the absolute amount and on the partitioning of the transported ruthenium between gaseous and aerosol fractions.
Addition of NO2 in a concentration of 50 ppmV into the gas stream significantly increased the gaseous fraction of ruthenium transported through the facility at all experimental temperatures. The overall transport of ruthenium was increased when compared to the humid air atmosphere at 1300 and 1700 K when NO2 was injected into the gas stream.
The number concentration of particles at the outlet of the facility was low in the case of NO2 feed, but the diameter of particles seemed to increase over the course of the experiments. These observations indicated the likelihood that part of the formed gaseous Ru compounds condensed on the surface of the existing particles and thereby increased the particle diameter.
Introduction of 50 ppmV N2O into the gas phase led to an increased fraction of ruthenium transported in the form of aerosols. The gaseous fraction of transported ruthenium was increased under all experimental temperatures. A very significant (almost 100%) increase in total transported ruthenium was observed in the experiment conducted at 1500 K when compared to the humid air atmosphere experiments.
With the injection of 5 ppmV HNO3 into the gas stream the transport of gaseous ruthenium increased at all studied temperatures. The overall transport of ruthenium with HNO3 in the air-flow was similar to that observed in the humid air atmosphere.
The examination of aerosols collected from the experiments by means of XPS and XRD techniques showed the same chemical speciation (anhydrous RuO2) over the entire range of experimental conditions.
The results obtained in this study showed a significant effect of nitrogen compounds on the transport of ruthenium in the primary circuit conditions. Introduction of nitrogen oxides and nitric acid into the gas stream promoted the transport of ruthenium tetroxide through the primary circuit simulating facility. The data obtained during this study provide additional insight into the ruthenium chemistry during a nuclear accident and reveal the possible interactions of ruthenium with air radiolysis products.
References
Haste T, Payot F, Bottomley PDW (2013) Transport and deposition in the PHÉBUS FP circuit. Ann Nucl Energy 61:102–121. doi:10.1016/j.anucene.2012.10.032
Grégoire AC, Haste T (2013) Material release from the bundle in PHÉBUS FP. Ann Nucl Energy 61:63–74. doi:10.1016/J.Anucene.2013.02.037
Ducros G, Pontillon Y, Malgouyres PP (2013) Synthesis of the VERCORS experimental programme: separate-effect experiments on fission product release, in support of the PHEBUS-FP programme. Ann Nucl Energy 61:75–87. doi:10.1016/j.anucene.2013.02.033
Bale C, Chartrand P, Degterov SA, Eriksson G, Hack K, Ben Mahfoud R, Melancon J, Pelton AD, Petersen S (2002) FactSage thermochemical software and databases. CALPHAD Comput Coupling Phase Diagr Thermochem 26(2):189–228. doi:10.1016/S0364-5916(02)00035-4
Eichler B, Zude F, Fan W, Trautmann N, Herrmann G (1992) Volatilization and deposition of ruthenium oxides in a temperature gradient tube. Radiochim Acta 56(3):133–140
Mun C, Cantrel L, Madic C (2007) Study of RuO4 decomposition in dry and moist air. Radiochim Acta 95(11):643–656. doi:10.1524/Ract.2007.95.11.643
Kärkelä T, Backman U, Auvinen A, Zilliacus R, Lipponen M, Kekki T, Tapper U, Jokiniemi J (2007) Experiments on the behaviour of ruthenium in air ingress accidents—final report. VTT Technical Research Centre of Finland, VTT research report—VTT-R-01252-07, Espoo
Kärkelä T, Vér N, Haste T, Davidovich N, Pyykönen J, Cantrel L (2014) Transport of ruthenium in primary circuit conditions during a severe NPP accident. Ann Nucl Energy 74:173–183. doi:10.1016/j.anucene.2014.07.010
Vér N, Matus L, Pintér A, Osán J, Hózer Z (2012) Effects of different surfaces on the transport and deposition of ruthenium oxides in high temperature air. J Nucl Mater 420(1–3):297–306. doi:10.1016/j.jnucmat.2011.09.030
Di Lemma FG, Colle JY, Beneš O, Konings RJM (2015) A separate effect study of the influence of metallic fission products on CsI radioactive release from nuclear fuel. J Nucl Mater 465:499–508. doi:10.1016/j.jnucmat.2015.05.037
Bosland L, Funke F, Langrock G, Girault N (2011) PARIS project: radiolytic oxidation of molecular iodine in containment during a nuclear reactor severe accident: Part 2. Formation and destruction of iodine oxides compounds under irradiation—experimental results modelling. Nucl Eng Des 241(9):4026–4044. doi:10.1016/j.nucengdes.2011.06.015
Vér N, Matus L, Kunstár M, Osán J, Hózer Z, Pintér A (2010) Influence of fission products on ruthenium oxidation and transport in air ingress nuclear accidents. J Nucl Mater 396(2–3):208–217. doi:10.1016/j.jnucmat.2009.11.008
Kajan I, Kärkelä T, Tapper U, Johansson L-S, Gouëllo M, Ramebäck H, Holmgren S, Auvinen A, Ekberg C (2017) Impact of Ag and NOx compounds on the transport of ruthenium in the primary circuit of nuclear power plant in a severe accident. Ann Nucl Energy 100(Part 2):9–19. doi:10.1016/j.anucene.2016.10.008
Backman U, Lipponen M, Auvinen A, Jokiniemi J, Zilliacus R (2004) Ruthenium behaviour in severe nuclear accident conditions—final report. VTT Technical Research Centre of Finland, VTT research report PRO3/P27/04, Espoo
Backman U, Lipponen M, Auvinen A, Tapper U, Zilliacus R, Jokiniemi JK (2005) On the transport and speciation of ruthenium in high temperature oxidising conditions. Radiochim Acta 93(5):297–304. doi:10.1524/Ract.93.5.297.64280
Mun C, Cantrel L, Madic C (2006) Review of literature on ruthenium behavior in nuclear power plant severe accidents. Nucl Technol 156(3):332–346
Roine A (2002) HSC chemistry for Windows 5.11, chemical reaction and equilibrium software with extensive thermochemical database, 5.11 edn. Outokumpu Research Oy, Pori
Erdtmann G (1976) Neutron activation tables. KErnchemie in Einzeldarstellungen, vol 6. Verlag Chemie, New York
JCGM 100:2008, evaluation of measurement data—guide to the expression of uncertainty in measurement JCGM 100:2008 (GUM 1995 with minor corrections) (2008). BIPM Joint Committee for Guides in Metrology, Paris
Kajan I, Lasseson H, Persson I, Ekberg C (2016) Interaction of ruthenium tetroxide with surfaces of nuclear reactor containment building. J Nucl Sci Technol 53(9):1397–1408. doi:10.1080/00223131.2015.1120245
Mun C, Ehrhardt J, Lambert J, Madic C (2007) XPS investigations of ruthenium deposited onto representative inner surfaces of nuclear reactor containment buildings. Appl Surf Sci 253(18):7613–7621. doi:10.1016/J.Apsusc.2007.03.071
Joint Committee on Powder Diffraction Standards (1970) Anal Chem 42(11):81A–81A. doi:10.1021/ac60293a779
Kärkelä T, Pyykönen J, Auvinen A, Jokiniemi J (2008) Analysis of flow fields, temperatures and ruthenium transport in the test facility. VTT Technical Research Centre of Finland, VTT-R-00947-08, NKS-161, Espoo
Polanyi JC (1956) Erratum: isotopic reaction rates between methyl and hydrogen. J Chem Phys 24(2):493–493. doi:10.1063/1.1742533
Huffman RE, Davidson N (1959) Shock waves in chemical kinetics: the thermal decomposition of NO 1a2 . J Am Chem Soc 81(10):2311–2316. doi:10.1021/ja01519a008
Harrison H, Johnston HS, Hardwick ER (1962) Kinetics of the thermal decomposition of nitric acid vapor. IV. A shock tube study between 800–1200°K. J Am Chem Soc 84(13):2478–2482. doi:10.1021/ja00872a004
Ellis WR, Murray RC (1953) The thermal decomposition of anhydrous nitric acid vapour. J Appl Chem 3(7):318–322. doi:10.1002/jctb.5010030706
Hinds WC (1999) Aerosol technology: properties, behavior, and measurement of airborne particles, vol Book, whole, vol 2. Wiley, New York
Holm J, Glänneskog H, Ekberg Ch (2009) Deposition of RuO4 on various surfaces in a nuclear reactor containment. J Nucl Mater 392:55–62
Fletcher JM, Jenkins IL, Lever FM, Martin FS, Powell AR, Todd R (1955) Nitrato and nitro complexes of nitrosylruthenium. J Inorg Nucl Chem 1(6):378–401. doi:10.1016/0022-1902(55)80048-6
Sasahira A, Kawamura F (1988) Formation rate of ruthenium tetroxide during nitric acid distillation. J Nucl Sci Technol 25(7):603–606. doi:10.1080/18811248.1988.9735899
Kim KS, Winograd N (1974) X-ray photoelectron spectroscopic studies of ruthenium-oxygen surfaces. J Catal 35(1):66–72
Ohyoshi A, Götzfried F, Beck W (1980) Polynuclear carbonyl complexes of ruthenium and osmium with methylthiolate and bromine bridging ligands. Chem Lett 9(12):1537–1540. doi:10.1246/cl.1980.1537
Acknowledgements
This study was performed as part of a Nordic Collaboration, ATR-2 (Impact of Aerosols on the Transport of Ruthenium) Experimental Programme, between Finland and Sweden. The financial support of SAFIR 2018, APRI 9 and NKS-R Programmes is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kajan, I., Kärkelä, T., Auvinen, A. et al. Effect of nitrogen compounds on transport of ruthenium through the RCS. J Radioanal Nucl Chem 311, 2097–2109 (2017). https://doi.org/10.1007/s10967-017-5172-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-017-5172-7