
Abstract
Resistance in Triticeae to fusarium head blight (FHB) is quantitatively inherited. Metabolomics as a tool was used to better understand the mechanisms of resistance and to identify potential FHB resistance biomarker metabolites in barley. Five FHB-resistant two-row barley genotypes (CIho 4196, Zhedar-1, Zhedar-2, Fredrickson, and Harbin-2r) and one FHB-susceptible genotype (CH 9520–30) were each inoculated with either pathogen-suspension or mock-solution. Disease severity, quantified as the proportion of spikelets diseased, varied among genotypes, being the greatest in CH 9520–30. Spikelets were sampled, metabolites extracted with aqueous methanol, and analyzed using an LC-ESI-LTQ-Orbitrap system. A pair wise, resistant vs. susceptible, t-test identified 1774 significant treatment peaks. Canonical discriminant analysis of peak abundance allowed the genotypes to be sorted into three clusters: (i) CH9520-30, (ii) Harbin-2r, (iii) the remaining four genotypes. The t-test was further used to identify resistance-related (RR) and pathogenesis-related (PR) metabolites. The pathogen-produced virulence factor deoxynivalenol (DON), and its detoxification product, DON-3-O-glucoside (D3G) were designated as resistance indicator (RI) metabolites. Metabolites (RR, PR, or RI) occurring in at least two resistant genotypes, showing a two-fold or greater abundance in resistant vs. susceptible lines, and also known to have plant defense functions were selected as potential FHB resistance biomarker metabolites. These included phenylalanine, p-coumaric acid, jasmonate, linolenic acid, total DON produced (TDP), and the proportion of DON converted to D3G (PDC). Total DON was the lowest in CIho 4196, while PDC was the highest in Zhedar-2. The application of RR, PR, and RI metabolites as potential biomarkers to enhance resistance is discussed.
Similar content being viewed by others
Introduction
Fusarium head blight (FHB) is a destructive disease of barley (Hordeum vulgare L.) and wheat (Triticum aestivum L.), with a worldwide economic impact (Gilbert and Tekauz, 2000). The disease is caused by several Fusarium species of which F. graminearum (teleomorph: Gibberella zeae) is of greatest relevance. Fusarium graminearum significantly reduces both grain yield and quality by producing trichothecene mycotoxins, such as deoxynivalenol (DON) (Desjardins 2006). Genetic improvement of host resistance to FHB is considered the best disease management option to reduce mycotoxin production and mitigate yield loss.
Plant resistance mechanisms to pathogen attack can be classified as apparent or true (Agrios, 2005). Apparent resistance is not directly controlled through a genomic influence on metabolism, but rather the plant escapes disease. For example, the closed-floret two-row barley genotypes are more resistant to infection than open-floret six-row genotypes (Bai and Shaner, 2004; Tekauz et al., 2000). True resistance is genetically controlled, and the mechanisms are either structural or biochemical, where the latter can be associated with particular proteins or metabolites. Quantitative FHB resistance documented in wheat and barley is associated with resistances to spikelet infection (Type I), spreading of the infection within the spike (Type II), and limiting trichothecene accumulation (Type III), especially DON, following pathogen invasion (Miller et al., 1986). While these categories of resistance have served in genotypic screening, they do not speak to the issue of resistance mechanisms. As mutants of F. graminearum lacking DON/trichothecene production fail to spread within infected spikes of wheat, DON is clearly an important virulence factor (Proctor et al., 1995; Jansen et al., 2005). In general, barley exhibits high type II resistance, therefore the focus of breeding programs has been on improving type I resistance (Bai and Shaner, 2004). However, ranking of barley genotypes based on type I resistance has proven to be inconsistent over years and seasons. This variation generally has been attributed to environmental conditions. More than 100 FHB-resistance quantitative trait loci (QTL) have been identified, across all seven chromosomes of wheat and barley, but several of these are either unstable or often associated with pleiotropic effects (Choo, 2006; Buerstmayr et al., 2009). The mechanisms of true resistance have been partially elucidated for a single QTL (chromosome 3BS), which confers a high rate of conversion of DON to DON-3-O-Glucoside (D3G) in resistant lines through the enzymatic action of DON-glucosyltransferase (Lemmens et al., 2005). The identification of FHB resistance mechanisms in barley would not only explain the functions of over 100 known FHB resistance QTLs but also would provide molecular breeders with novel tools for selecting resistant genotypes. In the present study, barley genotypes commonly used as sources of resistance in breeding and in QTL mapping programs were selected to explore and provide an overview of FHB resistance mechanisms controlled by metabolites.
Metabolomics technology has been applied to identify resistance-related (RR, greater abundance in resistant than in susceptible genotype) metabolites in wheat and barley. Based on gas chromatography and mass spectrometry (GC-MS), several RR metabolites have been identified (Hamzehzarghani et al., 2008). However, metabolomics based on GC-MS detect only volatile metabolites (Vorst et al., 2005). Based on LC-hybrid-MS (LC-ESI-LTQ-Orbitrap), more than 3000 peaks have been detected, and several RR metabolites have been identified in spikelets of barley in response to FHB infection (Bollina et al., 2010; Kumaraswamy et al., 2011). The metabolites identified belonged to three main chemical groups: phenylpropanoids, fatty acids, and terpenoids. While prior studies were based on an analysis of paired resistant/susceptible genotypes, in this study, the occurrence of RR metabolites was investigated in more than one resistant genotype, using an LC-MS method.
Methods and Materials
Plant Production
Five FHB-resistant two-row barley genotypes (CIho 4196, Fredrickson, Harbin-2r, Zhedar-1, and Zhedar-2) and one FHB-susceptible genotype (CH 9520–30) were used. Three seeds of each genotype were sown in individual 15 cm pots and maintained under greenhouse conditions at 22 ± 3°C. Plants were irrigated daily and fertilized at 2 wk intervals with 200 ml of 0.3% PlantProd (20-20-20 NPK + trace elements). Following germination, only the main stem and a single tiller of each plant were retained. These genotypes, along with several others, also were sown in a field in Ottawa, Ontario, Canada, as detailed in Choo et al. (2004). For each genotype, 10 g of seeds were sown in two rows of 1.5 m in length.
Pathogen Production, Inoculation, and Incubation
Fusarium graminearum Schwabe isolate 15–35 was maintained on potato dextrose agar media, and fresh cultures were produced on rye agar media. Seven-d old cultures were used to collect macroconidia. Barley spikelets were spray inoculated with 1 × 105 macroconidia ml-1 or an aqueous solution containing 0.02% Tween80 (mock), at 50% anthesis to early milky stage (GS = 65–73; Zadoks et al., 1974). The inoculated plants were placed inside plastic bags the inner surfaces of which had been sprayed with sterile water. The plants were maintained under these conditions for 48 h in order to facilitate initial establishment of the infection.
Experimental Design and Statistical Analyses
The greenhouse experiment was designed as a randomized complete block, with twelve treatments, consisting of six genotypes and two inoculations of mock-solution (M = 0.02% Tween80) or pathogen (P = macroconidia in 0.02% Tween80), with five replicates in time, conducted at roughly weekly intervals. Each experimental unit consisted of a pooled sample of 50–60 spikelets, collected from 6 plants. The field experiment was designed as a randomized complete block with four replicates (Choo et al., 2004). The experimental units consisted of 30 spikes per replicate. The resistance of the different genotypes was confirmed by assessing the proportion of spikelets diseased (PSD). The PSD was determined from the number of diseased spikelets in spray inoculated spikes 21 d after inoculation in the greenhouse or 1 mo after inoculation in the field.
Sampling and Metabolite Extraction
Some 50–60 inoculated spikelets, from 6 spikes, were harvested 72 h after inoculation. The lemmas and paleas were removed and frozen in liquid nitrogen, and metabolites were extracted (Kumaraswamy et al., 2011). In brief, the samples were crushed in liquid nitrogen with a mortar and pestle. A finely powdered 100 mg (fresh weight) sample was placed in a 2.2 ml micro-centrifuge tube, to which 1400 μl of ice-cold methanol (HPLC grade 99.96% pure, Fisher Scientific), 200 pg of genistein, and 200 pg of ribitol as internal standards, and 1,500 μl of water were added. After vortex mixing for 10 sec, the samples were sonicated for 15 min at a frequency of 40 kHz in a water bath at room temperature. The tubes then were centrifuged for 10 min at 20,000 g. The supernatants were filtered through a 0.1 μm PVDF membrane filter (Millipore Corporation, Bedford, MA, USA), and the filtrate was collected and stored at −20°C, and analyzed within a week.
Metabolite Analysis Using LC-ESI-LTQ-Orbitrap
Metabolites were analyzed using an LC-hybrid-MS system (LC-ESI-LTQ Orbitrap, Thermo Fisher, Waltham, MA, USA). Semipolar metabolites were analyzed with a capillary reverse phase C-18 column (500 μm i.d. × 10 cm ) packed with Jupiter 5 μm particle 300 Å reverse-phase stationary material (Phenomenex, Torrance) (Kumaraswamy et al., 2011). Polar metabolites, mainly trichothecenes from pathogen inoculated samples, were analyzed with a polar column, C18 Kinetex (210 μm i.d × 10 cm, with a particle size of 2.6 μm, Phenomenex). The column was maintained at 25°C, and the mobile phase was adjusted to a flow rate of 150 μl min−1 and eluted with water containing 2.5 mM ammonium acetate (A) and methanol (B). A gradient elution profile was used starting with 98% mobile phase A for 4 min, then changing to 10% mobile phase A over 26 min, and subsequently ramped to 90% mobile phase B over 5 min, followed by a 5 min ramp to 100% mobile phase B, and a rapid return to 100% mobile phase A. A blank sample was run after every experimental sample to reduce carry over. Some samples were analyzed in MS/MS mode, using 35 eV CID energy, to determine the fragmentation patterns. The compounds DON, 3ADON, and DON-3-O-glucoside (D3G) (Sigma-Aldrich, Oakville, ON, Canada) were spiked in different concentrations, using the above LC-hybrid-MS protocol. The abundances of DON, 3ADON, and D3G, rather than their acetate adducts, were used to develop regression models to predict their concentrations from abundances.
LC-MS Output Analysis
Raw data obtained from LTQ Orbitrap, based on semipolar and polar columns, were separately aligned using XCMS 1.22.1 (Kumaraswamy et al., 2011). Data were processed with a signal to noise threshold of, snthresh = 5:1 and band width of, bw = 10 s. The CAMERA bioinformatics tool (Tautenhahn et al., 2007) was used to annotate peaks with isotopes, neutral losses, adducts, and dimmers, which represented multiple peaks of the same compound. The data on peak abundance (relative ion counts) were subjected to a student’s t-test using SAS (version 9.2; SAS Institute Inc., Cary, NC, USA) to select metabolites that had significant treatment effects. The tests of hypotheses included four treatment combinations: RM vs. SM, RP vs. RM, SP vs. SM, RP vs. SP, where R = resistant, S = susceptible, M = mock, P = pathogen. The peaks with significant (P < 0.05) treatment effects were designated as metabolites.
The log transformed abundances of treatment significant metabolites, from mock-inoculated and pathogen-inoculated, were separately (because of the complex design) subjected to canonical discriminant analysis (CDA), to group observations, and identify the metabolites significantly loading to a group of treatments. The treatment significant metabolites also were further used to select pathogenesis related (PR), resistance related (RR), and resistance indicator (RI) metabolites. A metabolite with significantly higher abundance in a pathogen-inoculated than in a mock-inoculated genotype was considered a PR metabolite, be it in resistant (PRr = RP > RM) or susceptible (PRs = SP > SM) genotypes. A metabolite with a higher abundance in the resistant genotype than in a susceptible one was considered an RR metabolite. An RR metabolite based on mock-inoculation was considered as a constitutive (RRC = RM > SM) metabolite. A PRr metabolite in a resistant genotype with abundance greater than that in the susceptible pathogen-inoculated was considered as RR induced (RRI = RP > RM and RP > SP) metabolite (Hamzehzarghani et al., 2008). A relative reduction in the accumulation of the virulence factor DON (Proctor et al., 1995) in a resistant vs. susceptible plant served to indirectly assess two different plant resistance mechanisms: i) the reduction in DON synthesis by the pathogen, possibly due to high abundance of phenolics and flavonoids in the resistant genotype (Boutigny et al., 2009; Kushalappa lab unpublished data); or ii) the increased detoxification or conversion of DON to D3G, through the enzyme DON-glucosyltransferase (Lemmens et al., 2005; Bollina et al., 2010). The concentration of total DON produced was calculated as TDP = DON + D3G, using the regression models developed here to calculate concentrations from abundances. The proportion of TDP converted to D3G was calculated as: PDC = D3G/TDP. Both TDP and PDC were designated as resistance indicator (RI) metabolites. The PR, RR, and RI metabolites were assigned with putative names, based on accurate mass, fragmentation pattern that also considered manual verification, and isotope ratio, either matching with the spectra in databases or those obtained by spiking authentic standards (Bollina et al., 2010; Kumaraswamy et al., 2011).
Results
Disease Severity
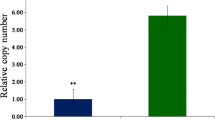
FHB severity (PSD), under greenhouse conditions, ranged from 0.54 in the susceptible genotype (CH 9520–30) to 0.17 in the most resistant genotype (CIho 4196). The PSD was not significantly different among five resistant genotypes tested, but was significantly lower in all resistant genotypes compared to the susceptible (Fig. 1). In field evaluations, the susceptible genotype CH 9520–30 had PSD of 0.48 as compared to 0.11 for CIho 4196. The ranking of six genotypes based on PSD in greenhouse evaluations were similar to those under field evaluations, except for Zhedar-1, which had higher PSD under field conditions.

Proportion of spikelets diseased in a spike (PSD), in six barley genotypes under greenhouse and field tests
Multivariate Analysis of Metabolic Profiles
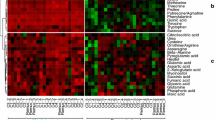
A total of 1774 metabolites had significant treatment effects. The abundances of the treatment-significant peaks from pathogen and mock-inoculated samples were separately subjected to canonical discriminant analysis (CDA) to better visualize the grouping of observations and genotypes. In both, the five replicates of different treatments were closely grouped, meaning there was little experimental error. In mock-inoculation (Fig. 2a), the CAN1 vector explained 60% of variance, with four resistant genotypes grouped together with high positive scores, and the fifth resistant genotype Harbin-2r as well as the susceptible genotype CH9520-30 both having negative scores. The genotypes CIho 4196 and Fredrickson had the highest positive scores. The metabolites with high positive loading to CAN1 vectors were: caffeoyl alcohol, kaempferol 7,4′-dirhamnoside, nallaflavanone, kaempferol 3-rhamnoside, linoleic acid, p-coumaric acid, and phenylalanine (Table S1a). In pathogen inoculated treatments (Fig. 2b), the CAN1 explained 41% of variance, where the resistant genotypes had high positive CAN1 scores, while the susceptible one had a highly negative CAN1 score. Although not all the high loading (L > 0.9) metabolites were identified, among those that were, octadecanoic and p-coumaric acids had relatively high loadings (Table S1b).

Canonical discriminant analyses (CDA) of metabolic profiles of six barley genotypes: a = metabolic profiles of pathogen inoculated; b = metabolic profiles of mock-inoculated
Resistance Related Constitutive Metabolites (RRC)
In total, 108, 84, 76, 106, and 69 RRC metabolites were detected in the genotypes CIho 4196, Zhedar-1, Zhedar-2, Fredrickson, and Harbin-2r, respectively (Table 1). Most of the RRC metabolites detected were phenylpropanoids. The magnitude of change in abundances of RRC metabolites relative to the susceptible variety varied among the resistant genotypes. A total of 14 RRC metabolites were common to two or more resistant genotypes, and their relative abundance was equal to or greater than two fold that in the susceptible genotype. The abundance of p-coumaric acid was 4.3 and 3.1 fold in Zhedar-2 and Fredrickson, respectively, while phenylalanine was nearly 2 fold in Zhedar-2, Zhedar-1, and Harbin-2r genotypes.
Pathogenesis Related (PR) and Resistance Related Induced Metabolites (RRI)
The genotypes CIho 4196, Zhedar-1, Zhedar-2, Fredrickson, and Harbin-2r had 12, 11, 26, 14, and 13 significantly induced metabolites (PRr), respectively, following pathogen inoculation. However, the amount of induction in resistant genotypes was not significantly higher than in susceptible genotypes, meaning that none of these were RRI metabolites. Jasmonic acid was a PRr metabolite in all five resistant genotypes. Linolenic acid was common to two resistant genotypes and two fold greater in abundance than in the susceptible genotype.
Resistance Indicator Metabolites (RI)
The extra cellular trichothecenes produced by the pathogen, DON, and 3ADON, and the plant enzymatic detoxification product of DON, D3G, were detected not only in their pure forms but also as their acetate (CH3COO−) adducts (Figs. S1, S2). The total amount of DON produced (TDP) by the pathogen, in lemma and palea, varied from 27 mg kg−1 in CIho 4196 to 53 mg kg−1 in Harbin-2r (Fig. 3a; Table 2). The amount of 3ADON, a precursor of DON, produced was quite low and it varied from 3.1 mg kg−1 in CH9520–30 to 4.4 mg kg−1 in Harbin-2r (Fig. 3a). The TDP was 50 mg kg−1 in susceptible genotypes CH9520–30, which was similar to the level in Harbin-2r. The PDC varied from 0.68 in CH9520–30 to 0.78 in Zhedar-2, and was moderate in Fredrickson, Harbin-2r, CIho 4196, Zhedar-1 (Fig. 3b; Table 2).

a Total DON produced (TDP = DON + D3G), DON-3-O-glucoside (D3G), and 3ADON in lemma + palea of six barley genotypes, following Fusarium graminearum inoculation, based on LC-ESI-LTQ-Orbitrap, in mg kg−1 of fresh weight of lemma and palea; b Proportion of TDP converted to D3G (PDC) in lemma and palea of six barley genotypes
PR, RR and RI Metabolites in Resistant Genotypes
CIho 4196
The most resistant genotype CIho 4196 had 109 RRC and 12 PRr metabolites. Several of the RRC metabolites showed many fold (×) changes in abundance, especially for conjugates of kaempferol (e.g., 2.0–7.6 fold changes). The changes in abundance of other RRC metabolites were: p-Coumaric acid = 2.8×, phenylalanine = 2.1×, linoleic acid = 3.8×, JA = 3.6×. Interestingly, the abundances of linoleic acid, linolenic acids, p-coumaric acid and phenylalanine increased more than two fold, following pathogen inoculation (PRr). The TDP was the lowest (27 mg kg−1) and the PDC was moderate (0.70), among the genotypes tested in this study (Table 2).
Zhedar-1
Had 84 RRC and 11 PRr meabolites (Table 1). The highest fold changes in abundance of RRC metabolites were observed for traumatic acid = 2.1×, linoleic acid = 1.9×, phenylalanine =2.2×, and eucommin A (lignan) = 3.2×. The changes for PRr metabolites were JA = 2.3× and stearic acid = 2.1×. The TDP was the second lowest (33 mg kg−1), but the PDC was quite low (0.70).
Zhedar-2
Had 76 RRC and 26 PRr metabolites. Large changes in abundance of RRC metabolites were observed for phenylalanine = 2.4×, p-coumaric acid = 4.3×, 4-geranyloxy-5-methylcoumarin = 2.3×, and eucommin A (lignan) = 1.4×, linolenic acid = 3.7×, and oleic acid = 1.9×. In addition, several flavonoids, as conjugates of kaempferol, were identified as RRC: 6-hydroxykaempferol 7,4′-dimethyl ether 3-sulfate (4.7×), kaempferol 7,4′-dirhamnoside (3.0×), kaempferide 5-glucoside-7-glucuronide (4.2×), kaempferol 3-(2″,3″-diacetyl-4″-p-coumarylrhamnoside (5.9×), and kaempferol 3-isorhamninoside (5.1×). The changes in abundance of PRr metabolites were: tuberculatin (lignan) = 1.3× and JA = 3.0×. The TDP was moderate (37 mg kg-1), but the PDC was the greatest among the genotypes evaluated (0.78) (Table 2).
Fredrickson
This genotype had 106 RRC and 14 PRr metabolites. Several phenylpropanoids and fatty acids were identified as RRC metabolites (Table 1). The changes in abundance were: p-coumaric acid = 3.1×, and an esterified coumaric acid, 4-geranyloxy-5-methyl coumarin = 2.7×. Other RRC metabolites were caffeoyl alcohol, tuberculatin (a lignan), traumatic acid, and stearic acid. The PRr metabolites were JA, linolenic acid, and heptadecatrienoic acid.
Harbin-2r
Had 69 RRC and 13 PRr metabolites. Among the ten RRC identified, four were phenylpropanoids and their change in abundance were: 4-geranyloxy-5-methyl coumarin = 3.2×, phenylalanine =2.0×, calophyllolide = 2.3×, and podorhizol beta-D-glucoside (lignan) = 1.7×. Among the four PRr metabolites identified, nortrachelogenin showd the greatest change (3.8×). The TDP was the highest (53 mg kg−1), but the PDC was the second highest (0.74) among genotypes tested (Table 2).
Discussion
Barley genotypes, with different levels of resistance to FHB, varied in their metabolic profiles. We have identified several resistance related constitutive metabolites. These RRC metabolites mainly belonged to three chemical groups: phenylpropanoids, flavonoids, and fatty acids. The occurrence of RRC metabolites, and their magnitude of change in abundance relative to a susceptible genotype, varied across resistant genotypes. The RRC metabolites that occurred in two or more resistant genotypes with at least two fold greater abundance relative to the susceptible were flavonoids: several conjugates of kaempferol, quercetin 3-(6″-acetylglucoside), triphyllin A, kuwanon L; phenylpropanoids: p-coumaric acid, phenylalanine, 4-geranyloxy-5-methyl coumarin, calophyllolide; fatty acids: linolenic acid and traumatic acid. Interestingly, several conjugates of kaempherol occurred in high abundance across different genotypes, with the highest increase relative to the susceptible genotype being 7.6× for CIho 4196. All the pathogen inoculated genotypes, not only accumulated DON but also detoxified DON to D3G, in lemma and palea. The most resistant genotype CIho 4196 had the lowest amount of TDP, along with the second highest PDC, the highest being in Zhedar-2 which had moderate TDP. Even though several pathogenesis related metabolites were detected in resistant genotypes, following pathogen inoculation, no resistance-related induced metabolite was identified. Jasmonic acid was a PRr metabolite in all the genotypes, with an up to 3.6× change in abundance, relative to the mock-inoculated. The present study indicates that barley has high levels of constitutive resistance against F. graminearum, as has been reported previously in other studies on wheat and barley (Hamzehzarghani et al. 2008; Bollina et al. 2010; Kumaraswamy et al. 2011). These metabolites can be used as potential biomarkers for screening genotypes for resistance against FHB. In addition to metabolites, proteins also significantly contribute to the biochemical resistance mechanisms of the plant against F. graminearum (Boddu et al. 2006; Zhou et al. 2006; Golkari et al. 2007; Geddes et al. 2008; Jia et al. 2009).
Metabolic profiling is still an evolving biochemical tool. Several solvents are needed to extract, and different platforms are needed to detect most of the metabolites in plant. Accordingly, we have not detected all the metabolites present in spikelets of the barley genotypes tested here. Using aqueous methanol for extraction and LC-hybrid-MS with a semipolar column (Jupiter) for analysis, we detected more than 1000 metabolites. Still, this column failed to consistently detect DON, 3ADON, and D3G, all of which were well-detected with a polar column (Kinetex). Furthermore, we failed to identify any RRI metabolites. This may be due mainly to spray inoculation of spikelets, where the variation in proportion of spikelets infected was high among replicates, which in turn can influence the abundance of metabolites. Individual inoculation of spikelets in wheat has yielded several RRI metabolites, including phenylpropanoids and fatty acids (Hamzehzarghani et al. 2008). In this study, only lemma and palea of the spikelets were analyzed, excluding the rest of the floral organs, as the lemma and palea are the primary defense structures enclosing the ovary/caryopsis, especially in two-row barley that has cleistogamous florets (Yoshida et al. 2007). Although Ma and Bohnert (2007) showed that each organ or tissue type had a specific transcriptome expression in the Arabidopsis genome, Golkari et al. (2007) found that the lemma, palea, and glume had similar gene expression patterns, but were different from rachis of wheat inoculated with F. graminearum.
Potential Biomarker Metabolites for FHB Resistance
RRC and RI Metabolites
The RRC and RI metabolites identified here, which met the following five criteria, were considered as potential biomarkers to screen barley genotypes for resistance against FHB: i) putatively identified (Table S2); ii) high fold change in abundance relative to susceptible; iii) highly significant, especially at P < 0.01 and P < 0.001 (Table S1); iv) occurred in more than one genotype; v) associated with plant defense function. Some of these metabolites, or precursors of these metabolites, also were identified as RR metabolites in our previous studies (Bollina et al. 2010; Kumaraswamy et al. 2011). Accordingly, these FHB resistance biomarker metabolites were grouped in their metabolic pathways.
Resistance Indicator Metabolites
Total DON Produced (TDP = DON + D3G)
In this study, the most resistant genotype CIho 4196 accumulated the lowest amount of TDP, and moderately resistant genotypes accumulated moderate amounts of TDP, except for Harbin-2r, which accumulated the highest amounts of TDP, similar to the susceptible CH9520-30 (Fig. 3a; Table 2). The accumulation of relatively lower amount of TDP by a resistant genotype can be due to the inhibition of DON/trichothecene synthesis, by the presence of high amounts of antioxidants, especially phenylpropanoids, as has been demonstrated for ferulic acid in vitro (Boutigny et al. 2009) and for others (Kushalappa et al. unpublished data). Ferulic acid represses the expression of several trichodiene synthase genes (Tri-genes), consequently inhibiting the synthesis of trichothecenes. The amount of DON accumulation in the plant at any time, depends on the amount of DON synthesized, less that converted to D3G. So, the determination of TDP, in this study, has enabled us to better evaluate the factors that affect DON synthesis. DON may not be needed for the initial invasion of spikelets (Jansen et al. 2005), but reduced synthesis of DON may reduce the rate of lesion expansion within florets. However, in our study, the reduced amounts of DON in resistant genotypes also could be due to reduced mycelial biomass relative to susceptible genotypes, even though the DON synthesis can be completely inhibited with little reduction in fungal biomass by phenolic compounds (Boutigny et al. 2009; Kushalappa et al. unpublished data).
Proportion of DON Conversion (PDC = D3G/TDP)
Even though RRI metabolites were not detected in our study, a resistance indicator (RI) metabolite, DON-3-O-glucoside, an enzymatic detoxification product of DON, by DON-glucosyltransferase, was detected (Bollina et al. 2010). DON is a virulence factor, and a mutant lacking trichothecene/DON synthesis failed to spread within a spike, in wheat (Proctor et al. 1995). In our study, all six barley genotypes converted DON to D3G, and PDC was the highest by Zhedar-2 (Table 2). Higher conversion ratios of up to 9:1 (D3G/DON) have been reported for wheat recombinant inbred lines with QTL at 3BS, which is associated with resistance to expansion of lesion to spikelets within spike (Lemmens et al. 2005). It is possible that the reduction in TDP and increase in PDC, in lemma and palea of resistant genotypes, as reported here, can lead to reduced expansion of lesion to spikelets through rachis. The ratio of D3G/DON may not correctly identify the mechanisms of resistance, as the DON reduction can be due to both DON conversion to D3G and reduced synthesis (Boutigny et al. 2009).
Phenylpropanoid Pathway
Phenylalanine
Phenylalanine, the precursor of the phenylpropanoid and flavonoid pathways, was detected as an RRC metabolite in Zhedar-1, Zhedar-2, and Harbin-2r. Although phenylalanine was not identified as an RR metabolite in our previous studies, several other metabolites related to the phenylalanine ammonia-lyase (PAL) pathway were (Bollina et al. 2010; Kumaraswamy et al. 2011). Although phenylalanine was detected in high amounts in Fredrickson, its level was not significantly different from that in the susceptible genotype. Two transcript-derived fragments TDF83 and TDF17 were strongly up-regulated upon Fusarium inoculation in FHB resistant genotypes, and both coded for phenylalanine ammonia-lyase (PAL) (Steiner et al. 2009).
p-Coumaric Acid
This was identified as an RRC in Zhedar-2 (4.3×) and Fredrickson (3.1×). It was a PRr metabolite in CIho 4196 (2.8×). p-Coumaric acid was identified as an RRC and PRr in our previous studies (Bollina et al. 2010; Kumaraswamy et al. 2011). It inhibited the production of biomass by F. graminearum on solid media, with an LD50 = 2.5 mM (Kumaraswamy et al. 2011), and an LD50 = 1.2 mM in liquid media (Bollina et al. 2010). It had a similar effect on both F. graminearum and F. culmorum at concentrations as low as 0.1 mg mL−1 in vitro (McKeehen et al. 1999). p-Coumaric acid accumulation also was associated with lignification of cell walls (Dixon 2001).
Flavonoid Pathway
Kaempferol
Several kaempferol conjugates were identified as RRC metabolites, with up to 7.6× changes in abundance (Table 1). The number of kaempferol conjugates was the highest in Zhedar-2. These also were observed among RRC in our previous studies (Bollina et al. 2010). Naringenin, a precursor of kaempferol, was identified as an RRC metabolite in our previous studies (Bollina et al. 2010; Kumaraswamy et al. 2011). Flavonoids are synthesized in the downstream phenylpropanoid metabolic pathway. The LD50 concentration of kaempferol was 4.8 mM on F. graminearum, as compared to 1.6 mM for its precursor metabolite naringenin (Bollina et al. 2010). However, the former was not completely soluble in aqueous methanol, thus its conjugates must be tested. Two other flavonoids with high fold change were identified: quercetin 3-(6″-acetylglucoside) (7.7×) and triphyllin A (8.2×), but these metabolites failed to meet all the five criteria used here to select biomarkers.
Fatty Acid Pathway
Jasmonic Acid
In this study, JA was identified as a PRr metabolite in all the five resistant genotypes. Even though it was not identified either as a RRC or as a RRI metabolite in this study (PRr not > PRs), it was considered here as a biomarker because of its importance as a signal molecule, especially in necrotrophic pathogenesis. Both JA and methyl-jasmonate (MeJA) were identified as PRr metabolites in our previous study, and the latter had LD50 = 1.0 mM for inhibition of mycelial growth on solid media (Kumaraswamy et al. 2011). Plant oxylipins, such as JA and MeJA are synthesized from polyunsaturated fatty acids (Shah 2009). Jasmonic acid and MeJA are the major signaling molecules in establishing resistance against necrotrophic pathogens (Robert-Seilaniantz et al. 2007). In wheat–F. graminearum interactions, a total of 14 transcripts were up-regulated, and they were considered to either encode JA biosynthetic enzymes or respond to JA (Jia et al. 2009). Two proteins, ankyrin repeat protein and 12-oxo-phytodienoic acid reductase, related to this signaling pathway were induced 3 days after F. graminearum inoculation, in wheat (Zhou et al. 2006). CIho 4196, showed an increased abundance for PR-9 proteins, indicating the possible use of JA antioxidant signaling pathway in pathogen defense (Geddes et al. 2008), which agrees with the results observed in this study.
Linoleic and Linolenic Acids
Zhedar-2 and Harbin-2r accumulated linolenic acid as an RRC metabolite. Linolenic acid, however, was also detected as a PRr metabolite in Fredrickson and CIho 4196. Linoleic acid, a precursor of linolenic acid, was identified as an RRC metabolite in Zhedar-1. Both linolenic and linoleic acids inhibited F. graminearum in vitro (LD50 = 3.6 mM for both, based on solid media) (Kumaraswamy et al. 2011). Linolenic acid is abundant in plasma membranes of wheat cells in the form of phospholipids (Li and Yen 2008), which serve as JA precursors. Linolenic acid can be released by the activities of a membrane-bound phospholipase, and then, oxidized to JA when the cells are under stress.
The potential biomarker metabolites for resistance in barley to F. graminearum established here can be used for screening barley genotypes for resistance against FHB, though further elucidation of the occurrence of these in other barley and wheat genotypes can better assess their suitability as biomarkers. Such studies also should identify more biomarkers, as in this study we failed to identify resistance-related induced metabolites, mainly because of high experimental error in spray inoculation. The monoisotopic accurate masses of these biomarkers can be selected in chromatography analyses to speed up the LC-hybrid-MS analysis and output process time. High throughput detection of several metabolites, based on direct infusion (metabolic fingerprinting), can be done in a few minutes. Alternatively, other cheaper tools such as HPLC can be standardized and used. Additionally, comprehensive proteomics can detect the enzymes involved in the biosynthesis of these biomarker metabolites, the sequence of which can be related to genes, which in turn, can be used in DNA marker assisted selections. Metabolic profiling tool, standardized here, can be applied to better understand mechanisms of multiple disease/stress resistance in plants.
References
Agrios, G. N. 2005. Plant Pathology. Elsevier Academic Press, London. p.922.
Bai, G., and Shaner, G. 2004. Management and resistance in wheat and barley to Fusarium head blight. Annu. Rev. Phytopathol. 42:135–161.
Buerstmayr, H., Ban, T., and anderson, J. 2009. QTL mapping and marker-assisted selection for Fusarium head blight resistance in wheat: a review. Plant Breeding 128:1–26.
Boddu, J., Cho, S., Kruger, W. M., and Muehlbauer, G. J. 2006. Transcriptome analysis of the barley–Fusarium graminearum interaction. Mol. Plant-Microbe Interact.19: 407–417.
Bollina, V., Kumaraswamy, G. K., Kushalappa, A. C., Choo, T. M., Dion, Y., Rioux, S., Faubert, D., and Hamzehzarghani, H. 2010. Mass spectrometry-based metabolomics application to identify quantitative resistance-related metabolites in barley against Fusarium head blight. Mol. Plant Pathol.11:769–782.
Boutigny, A., Barreau, C., Atanasova-Penichon, V., Verdal-Bonnin, M., Pinson-Gadais, L., and Richard-Forget, F. 2009. Ferulic acid, an efficient inhibitor of type B trichothecene biosynthesis and Tri gene expression in Fusarium liquid cultures. Mycol. Res. 113:746–753.
Choo T. M. 2006. Breeding barley for resistance to fusarium head blight and mycotoxin accumulation. Plant Breed Rev. 26:125–169.
Choo T. M., Vigier B., Chen Q. Q., Martin R. A., Ho K. M., and Savard M. 2004. Barley traits associated with resistance to fusarium head blight and deoxynivalenol accumulation. Phytopathology 94:1145–1150.
Desjardins, A. E. 2006. Fusarium Mycotoxins: Chemistry, Genetics and Biology, APS Press, St. Paul Minnesota, USA.
Dixon, R. A. 2001. Natural products and plant disease resistance. Nature 411:843–847.
Geddes, J., Eudes, F., Laroch, A., and Selinger, L. B. 2008. Differential expression of proteins in response to the interaction between the pathogen Fusarium graminearum and its host, Hordeum vulgare. Proteomics 8:545–554.
Gilbert, J. and Tekauz, A. 2000. Review: Recent developments in research on fusarium head blight of wheat in Canada. Can. J. Plant Pathol. 22:1–8.
Golkari, S., Gilbert, J., Prasher, S., and Procunier, J. D. 2007. Microarray analysis of Fusarium graminearum-induced wheat genes: identification of organ specific and differentially expressed genes. Plant Biotechnol. J. 5:38–49.
Hamzehzarghani, H., Paranidharan, V., Abu-Nada, Y., Kushalappa, A. C., Dion, Y., Rioux, S., Comeau, A., Yaylayan, V., and Marshall, W. 2008. Metabolic profiling coupled with statistical analyses for potential high throughput screening of quantitative resistance to fusarium head blight in wheat cultivars. Can. J. Plant Pathol.30:24–36.
Jansen, C., Von-Wettstein, D., Schäfer, W., Kogel, K., Felk, A., and Maier, F. 2005. Infection patterns in barley and wheat spikes inoculated with wild-type and trichodiene synthase gene disrupted Fusarium graminearum. Proc. Natl. Acad. Sci. USA 102:16892–16897.
Jia, H., Cho, S. and Muehlbauer, G. 2009. Transcriptome analysis of a wheat near-isogenic line pair carrying Fusarium head blight-resistant and-susceptible alleles. Mol. Plant-Microbe Interact. 22:1366–1378.
Kumaraswamy, G. K., Bollina, V., Kushalappa, A. C., Choo, T. M., Dion, Y., Rioux, S., Mamer, O., and Faubert, D. 2011. Metabolomics technology to phenotype resistance in barley against Gibberella zeae. Euro. J. Plant Pathol. 130:29–43.
Lemmens, M., Scholz, U., Berthiller, F., dall’asta, C., Koutnik, A., Schuhmacher, R., Adam, G., Buerstmayr, H., Mesterházy, Á., and Krska, R. 2005. The ability to detoxify the mycotoxin deoxynivalenol colocalizes with a major quantitative trait locus for fusarium head blight resistance in wheat. Mol. Plant-Microbe Interact.18:1318–1324.
Li, G. and Yen, Y. 2008. Jasmonate and ethylene signaling pathway may mediate Fusarium head blight resistance in wheat. Crop Sci. 48:888–896.
Ma, S. and Bohnert, H. 2007. Integration of Arabidopsis thaliana stress-related transcript profiles, promoter structures, and cell-specific expression. Genome Biol. 8:R49.
Mckeehen, J. D., Busch, R. H., and Fulcher, R. G. 1999. Evaluation of wheat (Triticum aestivum L.) phenolic acids during grain development and their contribution to Fusarium resistance. J. Agric. Food Chem. 47:1476–1482.
Miller, J. D., Young, J. C., and Arnison, P. G. 1986. Detoxification of deoxynivalenol by suspension cultures of a Fusarium head blight resistant wheat cultivar. Can. J. Plant Pathol. 8:147–50.
Proctor, R. H., Hohn, T. M., and Mccormick, S. P. 1995. Reduced virulence of Gibberella zeae caused by disruption of a trichothecene toxin biosynthetic gene. Mol. Plant-Microbe Interact. 8:593–601.
Robert-Seilaniantz, A., Navarro, L., Bari, R., and Jones, J. D. G. 2007. Pathological hormone imbalances. Curr. Opin. Plant Biol. 10:372–379.
Shah, J. 2009. Plants under attack: systemic signals in defense. Curr. Opin. Plant Biol. 12:459–464.
Steiner, B., Kurz, H., Lemmens, M., and Buerstmayr, H. 2009. Differential gene expression of related wheat lines with contrasting levels of head blight resistance after Fusarium graminearum inoculation. Theor. Appl. Genet. 118:753–764.
Tautenhahn, R., Böttcher, C., and Neumann. S. 2007. Annotation of LC/ESI-MS mass signals. Proceedings of BIRD 2007 - 1st International Conference on Bioinformatics Research and Development, Springer LNBI 4414.
Tekauz, A., Mccallum, B., and Gilbert, J. 2000. Review: Fusarium head blight of barley in western Canada. Can. J. Plant Pathol. 22:9–16.
Vorst, O., De Vos, C. H. R., Lommen, A., Staps, R. V., Visser, R. G. F., Bino, R. J., and Hall, R. D. 2005. A non-directed approach to the differential analysis of multiple LC-MS derived metabolic profiles. Metabolomics 1:169–180.
Yoshida, M., Kawada, N., and Nakajima, T. 2007. Effect of infection timing on Fusarium head blight and mycotoxin accumulation in open-and closed-flowering barley. Phytopathology 97:1054–1062.
Zadoks, J. C., Chang, T. T., and Konzak, B. F. 1974. A decimal code for the growth stages of cereals. Weed Res. 14:415–421.
Zhou, W., Eudes, F., and Laroche, A. 2006. Identification of differentially regulated proteins in response to a compatible interaction between the pathogen Fusarium graminearum and its host, Triticum aestivum. Proteomics 6:4599–4609.
Acknowledgements
This project was funded by the Ministère de l’Agriculture, des Pêcheries et de l’Alimentation du Québec (MAPAQ), Centre de recherche sur les grains inc. (CEROM), and the Fédération des producteurs de porc du Québec (FPPQ), Québec, Canada.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Supplementary Table S1
Phenylpropanoid, flavonoid and fatty acid RR metabolites identified in five Fusarium head blight (FHB) resistant barley genotypes, as compared to susceptible CH 9520–30 (DOC 80 kb)
Supplementary Table S2
Fragmentation patterns of biomarkers identified in barley genotypes against Fusarium head blight (FHB) (DOC 37 kb)
Figure S1
DON and its adduct, DON + CH3COO−, 3ADON and its adduct 3ADON + CH3COO−, and DON-3-O-glucoside (D3G) and and its adduct, D3G + CH3COO-, each of the three at one retention time (RT) (PPT 677 kb)
Figure S2
Abundances of DON, D3G, and 3ADON and their adducts in barley based on LC-LTQ-Orbitrap: a) Abundances of DON and DON + CH3COO−; b) Abundances of D3G and D3G + CH3COO−; c) Abundances of 3−ADON and 3-ADON + CH3COO− in lemma + palea of six barley genotypes inoculated with F. graminearum under greenhouse conditions, based on LC-ESI-LTQ-Orbitrap analysis (PPT 130 kb)
Rights and permissions
About this article
Cite this article
Kumaraswamy, K.G., Kushalappa, A.C., Choo, T.M. et al. Mass Spectrometry Based Metabolomics to Identify Potential Biomarkers for Resistance in Barley against Fusarium Head Blight (Fusarium graminearum). J Chem Ecol 37, 846–856 (2011). https://doi.org/10.1007/s10886-011-9989-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10886-011-9989-1