
Abstract
Chronic granulomatous disease (CGD) is the prototypic functional neutrophil disorder caused by genetic defects in one of the five genes encoding the superoxide-generating nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase subunits of phagocytes. Mutations causing the most prevalent form of CGD in western populations are located in the X-linked-CYBB gene. The four remaining autosomal recessive (AR) forms collectively account for one-third of CGD cases. We investigated the clinical and molecular features of eleven patients with CGD from 6 consanguineous families, originating from contiguous regions in the west of Tunisia. The patients’ clinical phenotype is characterized by a high incidence of mycobacterial infections. Five out of the eleven patients died despite treatment arguing in favor of a severe clinical form of CGD. These findings correlated with the absence of functional p67phox protein as well as the absence of residual reactive oxygen intermediates (ROI) production. Genetic analysis showed the presence, in all patients, of a unique mutation (c.257 + 2T > C) in NCF2 gene predicted to affect RNA splicing. Segregating analysis using nine polymorphic markers overlapping the NCF2 gene revealed a common haplotype spanning 4.1 Mb. The founder event responsible for this mutation was estimated to have arisen approximately 175 years ago. These findings will facilitate the implementation of preventive approaches through genetic counseling in affected consanguineous families.



Similar content being viewed by others
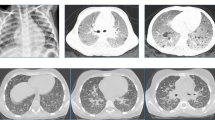
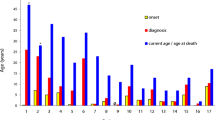

References
Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11.
van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic granulomatous disease: the European experience. PloS One. 2009;4(4):e5234.
Winkelstein JA, Marino MC, Johnston Jr RB, Boyle J, Curnutte J, Gallin JI, et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine. 2000;79(3):155–69.
Roos D, Kuhns DB, Maddalena A, Bustamante J, Kannengiesser C, de Boer M, et al. Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol Dis. 2010;44(4):291–9.
Lapouge K, Smith SJ, Groemping Y, Rittinger K. Architecture of the p40-p47-p67phox complex in the resting state of the NADPH oxidase. A central role for p67phox. J Biol Chem. 2002;277(12):10121–8.
Groemping Y, Rittinger K. Activation and assembly of the NADPH oxidase: a structural perspective. Biochem J. 2005;386(Pt 3):401–16.
Bylund J, Brown KL, Movitz C, Dahlgren C, Karlsson A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: how, where, and what for? Free Radic Biol Med. 2010;49(12):1834–45.
Babior BM. NADPH oxidase: an update. Blood. 1999;93(5):1464–76.
Kuijpers T, Lutter R. Inflammation and repeated infections in CGD: two sides of a coin. Cellul Molecul Life Sci CMLS. 2012;69(1):7–15.
Roos D, Kuhns DB, Maddalena A, Roesler J, Lopez JA, Ariga T, et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol Dis. 2010;45(3):246–65.
Matute JD, Arias AA, Wright NA, Wrobel I, Waterhouse CC, Li XJ, et al. A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood. 2009;114(15):3309–15.
Barbouche MR, Galal N, Ben-Mustapha I, Jeddane L, Mellouli F, Ailal F, et al. Primary immunodeficiencies in highly consanguineous North African populations. Ann N Y Acad Sci. 2011;1238:42–52.
Fattahi F, Badalzadeh M, Sedighipour L, Movahedi M, Fazlollahi MR, Mansouri SD, et al. Inheritance pattern and clinical aspects of 93 Iranian patients with chronic granulomatous disease. J Clin Immunol. 2011;31(5):792–801.
Koker MY, Camcioglu Y, van Leeuwen K, Kilic SS, Barlan I, Yilmaz M, et al. Clinical, functional, and genetic characterization of chronic granulomatous disease in 89 Turkish patients. J Allerg Clin Immunol. 2013;132(5):1156–63 e5.
Kim YM, Park JE, Kim JY, Lim HK, Nam JK, Cho M, et al. Genetic analysis of 10 unrelated Korean families with p22-phox-deficient chronic granulomatous disease: an unusually identical mutation of the CYBA gene on Jeju Island, Korea. J Korean Med Sci. 2009;24(6):1045–50.
Al-Zadjali S, Al-Tamemi S, Elnour I, AlKindi S, Lapoumeroulie C, Al-Maamari S, et al. Clinical and molecular findings of chronic granulomatous disease in Oman: family studies. Clin Genet. 2015;87(2):185–9.
Roesler J, Curnutte JT, Rae J, Barrett D, Patino P, Chanock SJ, et al. Recombination events between the p47-phox gene and its highly homologous pseudogenes are the main cause of autosomal recessive chronic granulomatous disease. Blood. 2000;95(6):2150–6.
Tanugi-Cholley LC, Issartel JP, Lunardi J, Freycon F, Morel F, Vignais PV. A mutation located at the 5′ splice junction sequence of intron 3 in the p67phox gene causes the lack of p67phox mRNA in a patient with chronic granulomatous disease. Blood. 1995;85(1):242–9.
Nathan DG, Baehner RL, Weaver DK. Failure of nitro blue tetrazolium reduction in the phagocytic vacuoles of leukocytes in chronic granulomatous disease. J Clin Invest. 1969;48(10):1895–904.
Genin E. Estimating the age of rare disease mutations: the example of triple-A syndrome. J Med Genet. 2004;41(6):445–9.
Graham SM, Ahmed T, Amanullah F, Browning R, Cardenas V, Casenghi M, et al. Evaluation of tuberculosis diagnostics in children: 1. Proposed clinical case definitions for classification of intrathoracic tuberculosis disease. Consensus from an expert panel. J Infect Dis. 2012;205 Suppl 2:S199–208.
El Kares R, Barbouche MR, Elloumi-Zghal H, Bejaoui M, Chemli J, Mellouli F, et al. Genetic and mutational heterogeneity of autosomal recessive chronic granulomatous disease in Tunisia. J Hum Genet. 2006;51(10):887–95.
Anwar WA, Khyatti M, Hemminki K. Consanguinity and genetic diseases in North Africa and immigrants to Europe. Eur J Public Health. 2014;24 Suppl 1:57–63.
Ben Halim N, Ben Alaya Bouafif N, Romdhane L, Kefi Ben Atig R, Chouchane I, Bouyacoub Y, et al. Consanguinity, endogamy, and genetic disorders in Tunisia. J Community Genet. 2013;4(2):273–84.
Organization WH. Tuberculosis country profiles. Available from: http://www.who.int/tb/country/data/profiles/en/.
Conti F, Lugo Reyes SO, Blancas Galicia L, Hubeau M, He J, Aksu G, et al. Mycobacterial diseases in 71 patients with chronic granulomatous disease. J Allergy Clin Immunol. 2016 doi:10.1016/j.jaci.2015.11.041.
Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600–10.
de Boer M, Tzur S, van Leeuwen K, Dencher PC, Skorecki K, Wolach B, et al. A founder effect for p47(phox)Trp193Ter chronic granulomatous disease in Kavkazi Jews. Blood Cells Mol Dis. 2015;55(4):320–7.
Ben-Mustapha I, Ben-Farhat K, Guirat-Dhouib N, Dhemaied E, Largueche B, Ben-Ali M, et al. Clinical, immunological and genetic findings of a large Tunisian series of major histocompatibility complex class II deficiency patients. J Clin Immunol. 2013;33(4):865–70.
Ben-Mustapha I, Ben-Ali M, Mekki N, Patin E, Harmant C, Bouguila J, et al. A 1,100-year-old founder effect mutation in IL12B gene is responsible for Mendelian susceptibility to mycobacterial disease in Tunisian patients. Immunogenetics. 2014;66(1):67–71.
Ouadani H, Ben-Mustapha I, Ben-Ali M, Ben-Khemis L, Largueche B, Boussoffara R, et al. Novel and recurrent AID mutations underlie prevalent autosomal recessive form of HIGM in consanguineous patients. Immunogenetics. 2016;68(1):19–28. doi:10.1007/s00251-015-0878-6.
Acknowledgments
We would particularly like to thank the patients and their family members whose cooperation was essential for the collection of the data used in this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ben-Farhat, K., Ben-Mustapha, I., Ben-Ali, M. et al. A Founder Effect of c.257 + 2T > C Mutation in NCF2 Gene Underlies Severe Chronic Granulomatous Disease in Eleven Patients. J Clin Immunol 36, 547–554 (2016). https://doi.org/10.1007/s10875-016-0299-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-016-0299-9