
Abstract
Inspired by the successful application of the embedded cluster reference interaction site model (EC-RISM), a combination of quantum–mechanical calculations with three-dimensional RISM theory to predict Gibbs energies of species in solution within the SAMPL6.1 (acidity constants, pKa) and SAMPL6.2 (octanol–water partition coefficients, log P) the methodology was applied to the recent SAMPL7 physical property challenge on aqueous pKa and octanol–water log P values. Not part of the challenge but provided by the organizers, we also computed distribution coefficients log D7.4 from predicted pKa and log P data. While macroscopic pKa predictions compared very favorably with experimental data (root mean square error, RMSE 0.72 pK units), the performance of the log P model (RMSE 1.84) fell behind expectations from the SAMPL6.2 challenge, leading to reasonable log D7.4 predictions (RMSE 1.69) from combining the independent calculations. In the post-submission phase, conformations generated by different methodology yielded results that did not significantly improve the original predictions. While overall satisfactory compared to previous log D challenges, the predicted data suggest that further effort is needed for optimizing the robustness of the partition coefficient model within EC-RISM calculations and for shaping the agreement between experimental conditions and the corresponding model description.
Similar content being viewed by others


Introduction
For more than a decade the SAMPL blind prediction challenges (Statistical Assessment of Modeling of Proteins and Ligands) [1] represent an optimal testbed for evaluating and optimizing the performance of computational models to predict experimental reference data. Our group participated in the past in a number of challenges on small molecule physicochemical properties, starting with SAMPL2 on tautomerization free energies in water [2], SAMPL5 on cyclohexane-water distribution coefficients (log D7.4) [3], SAMPL6.1 on aqueous pKa values [4], and SAMPL6.2 on octanol–water partition coefficients (log P) [5]. The methodology employed throughout was the embedded cluster reference interaction site model (EC-RISM) developed by us on the basis of combining three-dimensional (3D) RISM theory [6,7,8] as a solvation model with quantum–mechanical (QM) calculations [9]. This computational model allows for the calculation of Gibbs energies of species in solution that can be combined in thermodynamic cycles to yield derived quantities such as the previous SAMPL challenge targets mentioned above. The challenges themselves triggered further development of the model in terms of identifying and optimizing methodical details throughout the history, as has been discussed in broad detail in a recent overview paper [10]. Briefly summarizing the key results, we expect a pKa accuracy on the order of 1 and octanol–water log P accuracy below 1 pK units. log D7.4 values at the pH given as subscript have only been computed thus far for cyclohexane-water distributions, yielding expected errors on the order of 2 pK units, despite considerably better performance of the underlying pKa and log P models. This finding even holds for a re-evaluation of the older SAMPL5 dataset with the most highly optimized EC-RISM setup, giving rise to speculations about fundamental inconsistencies of the computational representation of experimental reality [10]. These issues have not been resolved yet as related but methodically different QM-based log D models typically exhibit similar error margins.
The latest SAMPL7 physical property challenge [11] represents a continuous further development, as participants were this time asked to predict both aqueous pKa values similar to SAMPL6.1 and octanol–water partition coefficients, log P, as during SAMPL6.2. Both quantities could be combined in the usual way to compute log D7.4 values. Experimental reference data on these quantities have been provided after the submission deadline although these were not part of the challenge. Based on our earlier experiences we decided to essentially apply our established models from SAMPL6.1 and 6.2 [4, 5]. Slight variations to be described below were not projected to influence the expected performance. As will be demonstrated, the performance of the acidity model even surpassed expectations while the partition coefficient results were significantly worse than found before for both training and SAMPL6.2 test set data, merging to an overall still satisfactory result for log D7.4 predictions. This inspired us in the post-submission phase to generate a new set of conformations to be tested as a potential source of uncertainty. Results of the original submission and the variation including consensus values are discussed in the following, also in comparison with data from other participants who submitted both pKa and log P predictions.
Computational details
As the RISM solvation Gibbs energy parametrizations for water and octanol as well as the optimized pKa model were taken from previous SAMPL challenges [4, 5] (with one minor adjustment for octanol described below), we here focus on comparing the different schemes for generating conformations of the challenge compounds that had been employed in the past.
For the submission, the workflow originally developed during the SAMPL5 challenge was applied to all microstates, including the additional relevant microstates complementing the set during the submission phase [1, 3, 10]. For each individual microstate, 200 conformations were generated starting from the original structures with the EmbedMultipleConfs utility of RDKit [12, 13]. If the molecule contained fewer than 7 rotatable bonds only 50 conformations were generated instead to reduce the computational cost for compounds with less conformational degrees of freedom. All conformations generated this way were optimized using the antechamber tool of the Amber12 suite [14], parametrized with AM1-BCC charges and GAFF version 1.7 parameters for bonded and non-bonded terms [14,15,16,17]. Solvation in water and octanol was simulated using an ALPB implicit solvation model with dielectric constants of 78.5 for water and 9.86294 for octanol, yielding two separate sets of 50 or 200 conformations each [18]. After the optimization an energy-filtered structural root mean square differences (RMSD) based clustering was applied to reduce the number of conformations to a more manageable number. Structures with a force field energy 20 kcal mol−1 above the apparent global minimum structure of a given microstate were discarded, with the minimum structure seeding the first cluster. All other conformations were then compared to the minimum structure in the order of increasing force field energies by using the GetBestRMS function of RDKit to calculate the RMSDs. If a structure had an RMSD of less than 0.5 Å it was discarded, while structures with a larger RMSD were added as additional cluster representatives. The resulting cluster representatives were optimized quantum-chemically using the B3LYP/6–311 + G(d,p)/IEFPCM level of theory implemented in Gaussian 16 Rev. C.01 [19]. After the quantum-chemical optimization another purely RMSD-based clustering using a cutoff of 0.5 Å was employed to remove conformations that reached the same minima during optimization. Up to five conformations with the lowest quantum-chemical energy were used in EC-RISM calculations to determine the Gibbs energy in solution per microstate by computing a partition function average. The compounds’ microstate Gibbs energies in the respective solvents \(G_{t}^{{{\text{sol}}}}\) was computed with the approach used in the SAMPL6 log P challenge by taking the sum of the electronic energy of the polarized wave function \(E_{tc}^{{{\text{sol}}}}\) and the corrected excess chemical potential \(\mu_{{tc{\text{,corr}}}}^{{{\text{ex}}}}\) of all conformations c per microstate t as
with \(\beta = (RT)^{ - 1}\) representing an inverse temperature. Detailed descriptions of how the electronic energies and excess chemical potentials are calculated and the specific corrections used for water and octanol can be found in previously publicized works [3,4,5]. The partition coefficient then follows from
with
After the original submission, the conformer generation approach used during the SAMPL6 challenges was also applied to the microstates of the SAMPL7 challenge to investigate if another set of conformations yields different results [4, 5]. In this case we generated the initial structures for QM optimization by using a force field-based sampling procedure. Structures of each microstate were taken as SMILES strings provided by the organizers. The flipper utility that is part of Omega [20] was used to perform a full enumeration of stereoisomers (i.e. generation of both formal E/Z isomers in cases they were not specified in the SMILES string), and initial 3D coordinates were generated using Corina [21]. For compounds bearing a sulfoxide moiety, additional stereoisomers with inverted chirality at the sulfur atom were added manually. The subsequent conformational analysis of all states was performed using Maestro 12.5 and Macromodel 12.9 as included in the 2020–3 release of the Schrödinger software suite [22]. Default parameters were used unless stated otherwise. We used the mixed torsional/low-mode conformational search algorithm and employed the OPLS3 force field in conjunction with an implicit water model. Conformational search up to a maximum of 1000 steps was carried out with 100 steps per rotatable bond present in the microstate. For saving conformations an energy window of 5 kcal mol−1 was used and redundant conformations were eliminated based on a RMSD cutoff of 1.5 Å. All resulting microstate conformations were forwarded to QM-based geometry optimization on the B3LYP/6–311 + G(d,p)/IEFPCM level of theory, and again up to 5 highest-ranking (lowest free energy) structures were selected for further processing by EC-RISM. Unlike the RDKit-based workflow employed for submission where different conformational sets for water and octanol were obtained und reoptimized, the sampling approach yielded only one set of conformations representative for water while final structural ensembles again differed slightly between solvents due to IEFPCM optimization mimicking the respective water and octanol environments.
For the EC-RISM calculations similar settings and solvent susceptibilities to those used in the SAMPL6 log P challenge were employed here to calculate the Gibbs energies of the compounds in solution, with one minor adjustment already pointed out as a perspective in our SAMPL6.2 paper [5]. Here, the water-saturated octanol solvent susceptibility was generated using the experimental number densities of 1.3598·10–3 Å−3 for water and 3.65787·10–3 Å−3 for octanol sites, and a dielectric permittivity of 8.41. As discussed in the original paper this is not expected to lead to significant deviations from the original water-saturated octanol model. Parametrization results and slightly changed resulting parameters for correcting the RISM excess chemical potential are shown in Fig. S1 and Table S1 in Online Resource (OR) 1. The 3D RISM calculations were conducted utilizing the PSE-2 closure [23] for water and the PSE-1 (Kovalenko-Hirata) closure for octanol. The RISM equations were solved on a cubic periodic grid of fixed size consisting of 1283 grid points and 0.3 Å grid spacing. The partial molar volumes entering the free energy correction expression [5] were calculated with the experimental compressibility of 0.761·10–9 Pa−1 for octanol and the 1D RISM estimate of the isothermal compressibility of 0.717062·10–9 Pa−1 for water [18, 24] from the total correlation function route. All EC-RISM calculations were done using the MP2/6–311 + G(d,p) level of theory within Gaussian 09 Rev. E.01 [25] using exact electrostatics taken directly from the wave function [4]. As in previous works, a more recent version of Gaussian was used for optimizations to take advantage of performance improvements [3, 5].
Aqueous pKa values were calculated from the optimized model developed in our SAMPL6.1 publication [4] for each pair of microstates separated by one unit charge difference and transformed, along with tautomer Gibbs energy differences, to the standard reaction free energy format required by the organizers by referring to a specific microstate reference [11]. As will be shown elsewhere in the SAMPL7 overview paper [26], the transformation from microstate pKa values (or corresponding standard reaction free energies) to the macrostate pKa values is equivalent to the “state transition” (ST) formalism analyzed by us recently [27, 28], so these values were submitted along with the microstate standard reaction free energies from microstate-specific Gibbs energies calculated according to Eq. (1). In the following we also compare these results to the “partition function” (PF) approach [27] using the same input data for state Gibbs energies. Gas phase energies were not needed, neither for pKa nor for log P calculations, as these cancel exactly because the gas phase ensembles of compounds evaporating from the water or the octanol phases are identical [10]. Finally, log D7.4 predictions were derived from calculated pKa and log P data in the usual way [3, 10].
Results and discussion
General outline and pK a predictions
We not only present our own data but also try to put the results into context by comparison with other participants. Here we chose only those submissions for which the final quantity, log D7.4 could in principle be calculated, i.e. challenge contributions containing both, ranked pKa and log P predictions. Without going into methodical detail, the following 5 submissions satisfied the conditions, termed according to the submission nomenclature (1) “MD (CGenFF/TIP3P)|Gaussian_corrected”, (2) “TFE-SMD-solvent-opt|DFT_M06-2X_SMD_explicit_water”, (3) “TFE-NHLBI-TZVP-QM|TZVP-QM”, (4) “TFE IEFPCM MST|IEFPCM/MST”, (5) “TFE b3lypd3|DFT_M05-2X_SMD” [11]. The first part in front of the pipe symbol refers to the log P model, the second to the pKa approach. Accordingly, our own models are termed (0) “EC_RISM_wet|EC_RISM”. As outlined in the preceding section, besides data from the original structure set (“orig”) we also report results from the new set of geometries (“new”) separately and from a combination (“comb”) by simply augmenting the microstate partition function with the new energies, ignoring the possibility of duplicates. In the following analysis of acidity constants, the state transition approach [27] was used for deriving macroscopic pKa values from submitted free energies throughout for all submissions.
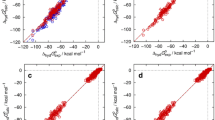
All pKa models agreed in the choice of the relevant ionization state change related to the observed macroscopic pKa values, going from a neutral acid to a negatively charged base, which greatly simplified the analysis. Transitions from charged acids were accompanied throughout by negative pKa predictions and could be ignored for comparison with experiment. Results for macroscopic acidity constants are shown in Table 1 and Fig. 1 with individual compound data summarized in Table 2. Apparently, EC-RISM outperformed other methods, exceeding expectations from earlier challenges and the training set performance (ca. 1 pK unit RMSE) with a submission RMSE of 0.72 pK units. High correlation measured by R2 and a regression slope near one, small absolute and signed errors indicate an overall robust model. The new set of conformations performed slightly inferior, though still in line with the metrics of the original set and not overlapping with prediction statistics of other models. Somewhat unexpectedly it turned out that the combined set of conformations did not lead to improvement. This means that the new conformations do not fully overlap with the old ones but add some new low-energy structures to the partition function that yield larger deviations in terms of their pKa performance. The only conclusion at this point is that the observed discrepancy between different conformation sets can be taken as a measure of model uncertainty (not to be confused with expected prediction uncertainty).

Macroscopic acidity constants for the SAMPL7 set calculated using EC-RISM (A) and other models discussed in this work (B) with different sets of conformations in panel (A) encoded by symbol and line colors, original: black, (0) EC_RISM (orig); new: orange, (0) EC_RISM (new); combined: green, (0) EC_RISM (comb). The model comparison in panel (B) is color-coded as black: (0) EC_RISM (orig), orange: (1) Gaussian_corrected, green: (2) DFT_M06-2X_SMD_explicit_water, yellow: (3) TZVP-QM, magenta: (4) IEFPCM/MST, blue: (5) DFT_M05-2X_SMD. Two data points of submission (2) DFT_M06-2X_SMD_explicit_water are not shown in panel (B) as they lie far outside the experimental range. Dashed lines indicate descriptive linear regression results. Raw data are provided as OR3 for structures and OR4 for energies. Macroscopic pKa values for other participants’ models were taken from the SAMPL7 repository [11] and are additionally collected in OR7
Individual compound data in Table 2 further illustrates the prediction balance, with the largest deviation between prediction and experiment on the order of 1.6 pK units found for SM34 and SM39. For completeness, we there also show results from applying the partition function approach [27] which – as expected – only marginally differs from the state transition results.
log P and log D 7.4 predictions
Given the successful application of the EC-RISM model to octanol–water phase partitioning of neutral compounds during SAMPL6.2 [5] (training and test set RMSEs of ca. 1.5 and 0.5 pK units), we expected similar performance for the SAMPL7 compound set. However, numbers reported in Tables 3 (statistical metrics) and 4 (individual compound data) and illustrated in Figs. 2 and 3 for log P and log D7.4, respectively, show a satisfactory, yet worse than expected overall result. With a log P RMSE for the original conformations of 1.84 pK units the upper limit of our expectation was slightly exceeded, and the non-zero MSE and regression intercept indicates a systematic trend to overestimate log P values, which has not been observed with our models before. Adding new conformations here somewhat improves the results, unlike the pKa case, but not to an extent that we would assume to have pinpointed the origin of the discrepancies. It is possible that the specific chemistry of the SAMPL7 set is so different from earlier datasets tested that our model development is not yet robust enough to capture very diverse systems. One candidate for deeper investigation is the element sulfur which is not well represented in our reference datasets and which could have implications for the chosen theoretical level of theory, most likely the basis set.

Partition coefficients for the SAMPL7 set calculated using EC-RISM (A) and other models discussed in this work (B) with different sets of conformations in panel (A) encoded by symbol and line colors, original: black, (0) EC_RISM_wet (orig); new: orange, (0) EC_RISM_wet (new); combined: green, (0) EC_RISM_wet (comb). The model comparison in panel (B) is color-coded as black: (0) EC_RISM_wet (orig), orange: (1) MD (CGenFF/TIP3P), green: (2) TFE-SMD-solvent-opt, yellow: (3) TFE-NHLBI-TZVP-QM, magenta: (4) TFE IEFPCM MST, blue: (5) TFE b3lypd3. Dashed lines indicate descriptive linear regression results. The signs of log P predictions for model (3) TFE-NHLBI-TZVP-QM have been inverted as accidentally the wrong reaction direction has been submitted. The log P values for other participants’ models were taken from the SAMPL7 repository [11]. Raw data are provided as OR5 for structures and OR6 for energies and are additionally collected in OR7

Distribution coefficients for the SAMPL7 set calculated using EC-RISM (A) and other models discussed in this work (B) with different sets of conformations in panel (A) encoded by symbol and line colors, original: black, (0) EC_RISM_wet (orig); new: orange, (0) EC_RISM_wet (new); combined: green, (0) EC_RISM_wet (comb). The model comparison in panel (B) is color-coded as black: (0) EC_RISM_wet (orig), orange: (1) MD (CGenFF/TIP3P)|Gaussian_corrected, green: (2) TFE-SMD-solvent-opt|DFT_M06-2X_SMD_explicit_water, yellow: (3) TFE-NHLBI-TZVP-QM|TZVP-QM, magenta: (4) TFE IEFPCM MST|IEFPCM/MST, blue: (5) TFE b3lypd3|DFT_M05-2X_SMD. Dashed lines indicate descriptive linear regression results. One data point of submission (2) TFE-SMD-solvent-opt|DFT_M06-2X_SMD_explicit_water is not shown in panel (B) as it lies far outside the experimental range. The signs of underlying log P predictions for model (3) TFE-NHLBI-TZVP-QM|TZVP-QM have been inverted as accidentally the wrong reaction direction has been submitted. Calculated data are provided as OR7
Compared to the other log P models analyzed in this work our results rank average, with the best performing model (4) yielding an RMSE of ca. 1 pK unit. However, all models analyzed, including our own, show very little degree of correlation measured by R2, despite relatively reasonable regression slopes. This can be clearly traced back to a number of substantial outliers (e.g. SM42, SM43, see Table 4), for which there is no apparent explanation. The RMSE-wise best model (4) yields even a smaller value for this metric than ours, hinting at the possibility that chance plays a large role for obtaining good results.
Results from log D7.4 predictions are slightly better, being even below our expectation of more than 2 pK units deviation with an RMSE of 1.69 pK units, ranking second (by a very small margin to the third) in the field of challenge participants with the best model (4) reaching 1.27. Here, adding new conformations again slightly worsened results due to weaker performance already observed for pKa values. Scatter is, however, still large, so it is not possible to draw some general performance conclusions for this small and chemically focused dataset. One trend is obvious: Physics-based models such as those analyzed and compared in this work, that perform reasonably well and balanced in different prediction domains, will also perform well in combined model problems such as log D7.4 predictions. Still, log D7.4 remains a challenging property to be examined further in order to understand and improve model weaknesses. There is also room for improvement on the experimental side. We noted in some cases (see Table 4) that originally measured and reconstructed log D7.4 from pKa and log P differ. Although there is apparently no correlation with prediction performance or failure, this could at least stimulate questions to further converge computational representations to match experimental reality.
Concluding discussion
The most remarkable finding in this work is that apparently different conformational search or sampling strategies even for rather small molecules like those of the SAMPL7 set yield quite different results. Time did not permit deeper analysis of individual conformations, but it is clear that extended effort is needed for developing more consistent conformational sampling workflows. It is very likely that the problem originates already from the initial force field sampling stage as further QM-based optimization including a solvation model did not yield converged conformational ensembles.
However, our results show that conformational uncertainties alone are not responsible for the observed errors in thermodynamic quantities, which in our case imply an overestimated hydrophobicity. For water, results appear to be more reliable than for octanol, despite our earlier findings during SAMPL6.1 and SAMPL6.2 from which we expected better performance for log P than for pKa predictions. In light of the different chemistry of SAMPL7 compared to SAMPL6 compounds, this hints at a possibly problematic description of sulfur-octanol interactions which could be related to the QM level of theory and/or sulfur-octanol dispersion interactions that are not modeled by first principle methods but by empirical Lennard–Jones terms. In the SAMPL7 challenge each compound contains a sulfone moiety whereas this functional group is represented by only one single MNSOL database entry, (sulfolane, test2027). This compound was predicted with an error of 4.83 kcal mol−1 for octanol, the largest in the entire training set [5]. For water the error is only 3.63 kcal mol−1, so it is likely that the error cancellation within the same solvent, as seen for the acid/base pair within pKa predictions, does no longer apply for transfer free energies between different solvents. However, more solvent-specific experimental data, such as solvation free energies are necessary to confirm this hypothesis.
Another remarkable observation is that log D values taken directly from experiment or from a reconstruction based on experimental acidity and partition coefficients do not yield identical numbers in all cases. In cases where the two approaches differ significantly, i.e. for SM25, 26, 41–43, the reconstructed distribution coefficient is smaller, i.e. more negative than the direct measurement. This means that possibly a higher amount of the compound is dissolved in the aqueous phase than expected from neutral state partitioning alone if we take the reconstructed data as correct. If we, however, accept the direct experimental result then the opposite conclusion would emerge, namely that a larger compound fraction is dissolved in the organic phase. In other words, this could be interpreted as a missing contribution of charged species in the organic phase in our calculations where, via the standard formula for converting log P to log D, the presence of charged microstates in the nonaqueous phase is by definition excluded. This statement should in any case be viewed with caution as a range of alternative explanations could come into play, such as aggregation, nonideality effects due to insufficient dilution and so forth. However, observed inconsistencies are again a source and stimulus of deeper analysis including the correct agreement between experimental reality and its computational model representation.
References
https://samplchallenges.github.io. Accessed 29 Mar 2021
Kast SM, Heil J, Güssregen S, Schmidt KF (2010) J Comput-Aid Mol Des 24:343–353
Tielker N, Tomazic D, Heil J, Kloss T, Ehrhart S, Güssregen S, Schmidt KF, Kast SM (2016) J Comput-Aid Mol Des 30:1035–1044
Tielker N, Eberlein L, Güssregen S, Kast SM (2018) J Comput-Aid Mol Des 32:1151–1163
Tielker N, Tomazic D, Eberlein L, Güssregen S, Kast SM (2020) J Comput-Aid Mol Des 34:453–461
Beglov D, Roux B (1997) J Phys Chem 101:7821–7826
Kovalenko A, Hirata F (1998) Chem Phys Lett 290:237–244
Sato H (2013) Phys Chem Chem Phys 15:7450–7465
Kloss T, Heil J, Kast SM (2008) J Phys Chem B 112:4337–4343
Tielker N, Eberlein L, Hessler G, Schmidt KF, Güssregen S, Kast SM (2020) J Comput-Aid Mol Des 35:453–472
https://github.com/samplchallenges/SAMPL7. Accessed 29 Mar 2021
RDKit: Open-source cheminformatics, https://www.rdkit.org. Accessed 29 Mar 2021
Ebejer J-P, Morris GM, Deane CM (2012) J Chem Inf Model 52:1146–1158
Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Hayik S, Roitberg A, Seabra G, Swails J, Götz AW, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wolf RM, Liu J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh MJ, Cui G, Roe DR, Mathews DH, Seeting MG, Salomon-Ferrer R, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA (2012) AMBER 12, University of California, San Francisco, USA, https://ambermd.org. Accessed 3 March 2021
Sigalove G, Fenley A, Onufriev A (2006) J Chem Phys 124:124902
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA (2004) J Comput Chem 25:1157–1174
Jakalian A, Jack DB, Bayly CI (2002) J Comput Chem 23:1623–1641
Lide DR (2004) CRC handbook of chemistry and physics, 84th edn. CRC Press, Boca Raton
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2016) Gaussian 16 Rev. C.01, Wallingford CT.
Hawkins PCD, Skillman AG, Warren GL, Ellingson BA, Stahl MT, OMEGA 2.6.7: OpenEye Scientific Software, Santa Fe, NM
3D Structure Generator CORINA Classic, version 4.1.0, Molecular Networks GmbH, Nuremberg, Germany
Small-Molecule Drug Discovery Suite 2020-3 (2020), Schrödinger, LLC, New York
Kast SM, Kloss T (2008) J Chem Phys 129:236101
Imai T, Kinoshita M, Hirata F (2000) J Chem Phys 112:9469–9478
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09 Rev. E.01, Wallingford CT.
Bergazin TD, Tielker N, Zhang Y, Mao J, Gunner MR, Francisco K, Ballatore C, Kast SM, Mobley DL (2021). J Comput-Aid Mol Des. https://doi.org/10.1007/s10822-021-00397-3
Tielker N, Eberlein L, Chodun C, Güssregen S, Kast SM (2019) J Mol Model 25:139
Bochevarov AD, Watson MA, Greenwood JR (2016) J Chem Theory Comput 12: 6001–6019
Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC-2033 – Projektnummer 390677874, and under the Research Unit FOR 1979. We also thank the IT and Media Center (ITMC) of the TU Dortmund for computational support. We would also like to express our gratitude to the organizers of the SAMPL challenges (current funding source NIH Grant R01GM124270) as well as to all producers of experimental reference data.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tielker, N., Güssregen, S. & Kast, S.M. SAMPL7 physical property prediction from EC-RISM theory. J Comput Aided Mol Des 35, 933–941 (2021). https://doi.org/10.1007/s10822-021-00410-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10822-021-00410-9